工場では実際にモノを作る前に,CAEによる論理シミュレーションが行われる(工場のしくみ,p.166).
分子軌道計算は,材料の反応予測や材料物性の予測に利用される.
プラントを建設する前に,論理シミュレータでプラントを設計する.
材料物性の予測・・・分子軌道計算。エネルギー最少。
ハードウェア込みで500万円超?、アカデミックディスカウント。
計算科学の歴史。1953年 MD、1973年、ガウシャンPDP-11、1985年80386DX。
電子出願ソフトサポートサイト
特許
の明細書に分子式を記述するときの
画像ファイル
の形式は?
検索
情報を表現しようとしたら、文字にしなければなりません。
デジタル化とはそういうことです。
全てを文字で表現することはできないし、すべてを文字で表現すべきでもありません。
文字にする価値が期待できる情報を文字にすべきです。
デジタルの世界では、
グラフィックスや
色彩、
音声まで文字で表現します。
化学式は、化学物質や反応を文字で表現したものと言えます。
しかし、そこには、下付き文字や上付き文字などが入ってきます。
まして構造式などになると、グラフィックスで表現せざるを得ません。
コンピュータに読み書きしやすい文字が、人間にとって必ずしも読み書きしやすいとは限りません。
表
2
.
表現のためのファイル形式の例
| 表現 | |
|---|
| |
|
|
◇バイナリ形式
任意のビット列
|
◇テキスト形式
文字コードのみ
|
| ネイティブ |
圧縮テキスト |
テキスト |
◇ XML
|
| 文書 |
doc |
docx |
◇html,html5 |
xhtml |
| 表計算 |
xls |
xlsx |
◇csv |
|
| 図形 |
◇MWF,EMF |
|
vml |
◇
svg |
| チャート |
|
|
UML |
|
| 地図 |
|
|
|
◇G-XML,GML
|
| 数式 |
マセマティカ
|
|
◇TEX |
◇ MathML |
| 化学式 |
|
|
SMILES |
CML |
| 楽譜 |
MIDI |
|
MML |
MusicXML |
| 3D |
|
|
VRML |
X3D |
|
画像
(image)
|
jpg,png |
|
|
|
| 音楽
(audio)
|
wav,mp3,wma |
|
|
|
|
ビデオ
(video)
|
mp4,mov |
|
|
|
| アプリ |
exe,dll |
|
vb,js,asp |
|
SMILES記法
International Chemical Identifier(InChI)
SMILES記法チュートリアル
ChemBioDraw/ChemDraw
BIOVIA Draw(バイオビアドロー)
ChemDraw
ChemDraw for iPad
1970年 Gussian登場, PDP-11,非経験的分子軌道法,ab initio分子軌道法
1983年 MOPACが登場, 半経験的分子軌道法
jsmolを使って表現した粘土
原子と原子は静電気力で結びつく。化学結合にはイオン結合、共有結合、金属結合などがある。
固体には、塩化ナトリウムなどのイオン結晶、
ダイヤモンドやケイ素などの共有結合からなる共有結晶、
銀などの金属結合からなる金属結晶、
供給結合からなる分子が弱い力でくっつくドライアイスなどの分子結晶などがある。
液体は水のように分子からなる液体、
塩化ナトリウムを高温でナトリウムイオンと塩化物イオンがイオン液体(熔融塩)、
銀が自由に動き回る溶融金属、などがある。
ドライアイスやヨウ素などの分子結晶は昇華しやすく、液体にはなりにくい。
電子軌道(オービタル)とエネルギー準位
表
3
.
電子軌道(オービタル)とエネルギー準位
|
電子軌道の種類
|
対象
|
軌道の数
|
フェルミ準位
|
|
|
|
|
|
|
|
原子軌道(AO)
|
s,p,d,f
|
原子に属する
電子の個数の半分程度
|
|
|
分子軌道(MO)
|
σ、π、δ
|
分子に属する
原子の電子の個数の半分程度
|
HOMO、LUMO
|
|
バンド
|
|
固体に属する原子の数(アボガドロ数)程度
|
価電子帯、導電帯
|
放電管に
水素をわずかに入れ(ガイスラー管程度、低真空=10−3 atm(100 Pa))放電すると、
水素原子特有の光を発します。
これを分光器で分光すると、
赤と
シアンの
線スペクトルが観察されます。
これは、水素原子の中の電子が、とびとびのエネルギーしか許されないことを表しています。
有機化学美術館
分子全体に広がる一電子空間軌道関数である分子軌道によって、分子を構成する個々の電子の状態が記述される。
この分子軌道を計算して、分子の電子状態を求める方法が分子軌道法である。コンピュータで計算する手法である.なお,分子軌道法については,物理化学IIIの授業で詳しく触れます.
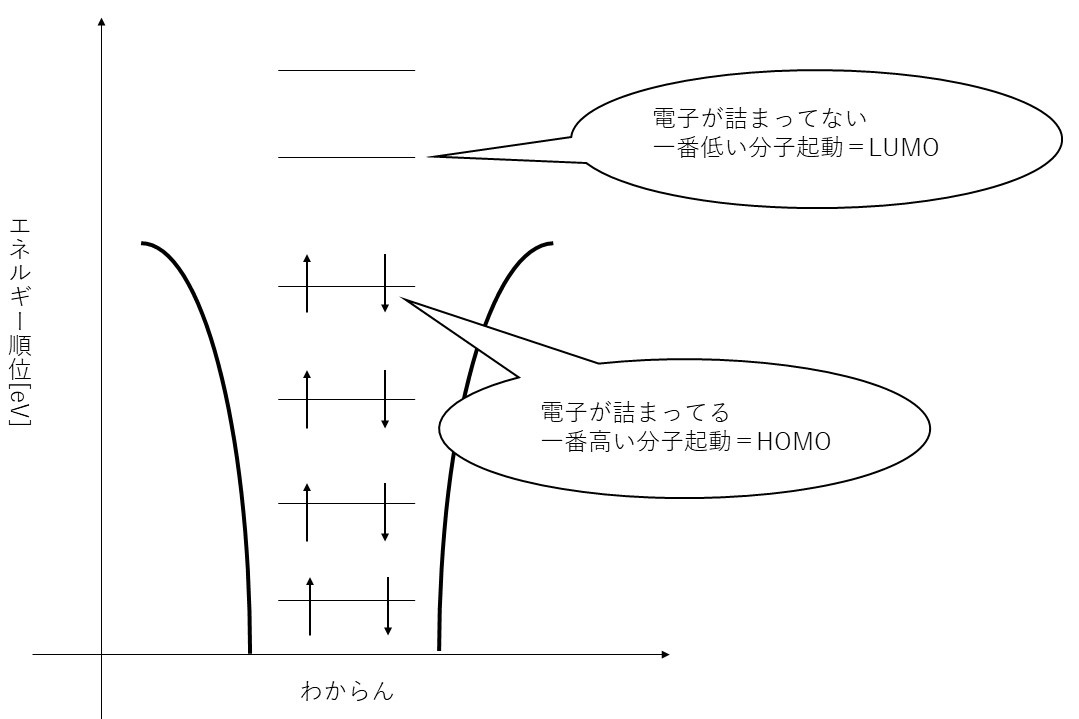 図 144 クーロンポテンシャルとHOMOとLUMO
図 144 クーロンポテンシャルとHOMOとLUMO
©Copyright Takaaki Shiraya all rights reserved.
上図を3Dで可視化したものが下図です。
クーロンポテンシャルは原子核のまわりにできるへこみみたいなものです。
電子はそこにたまる水みたいなものです。
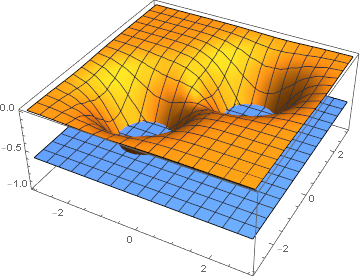 Fig
クーロンポテンシャルとHOMOのイメージ(
マセマティカによる可視化)
Fig
クーロンポテンシャルとHOMOのイメージ(
マセマティカによる可視化)
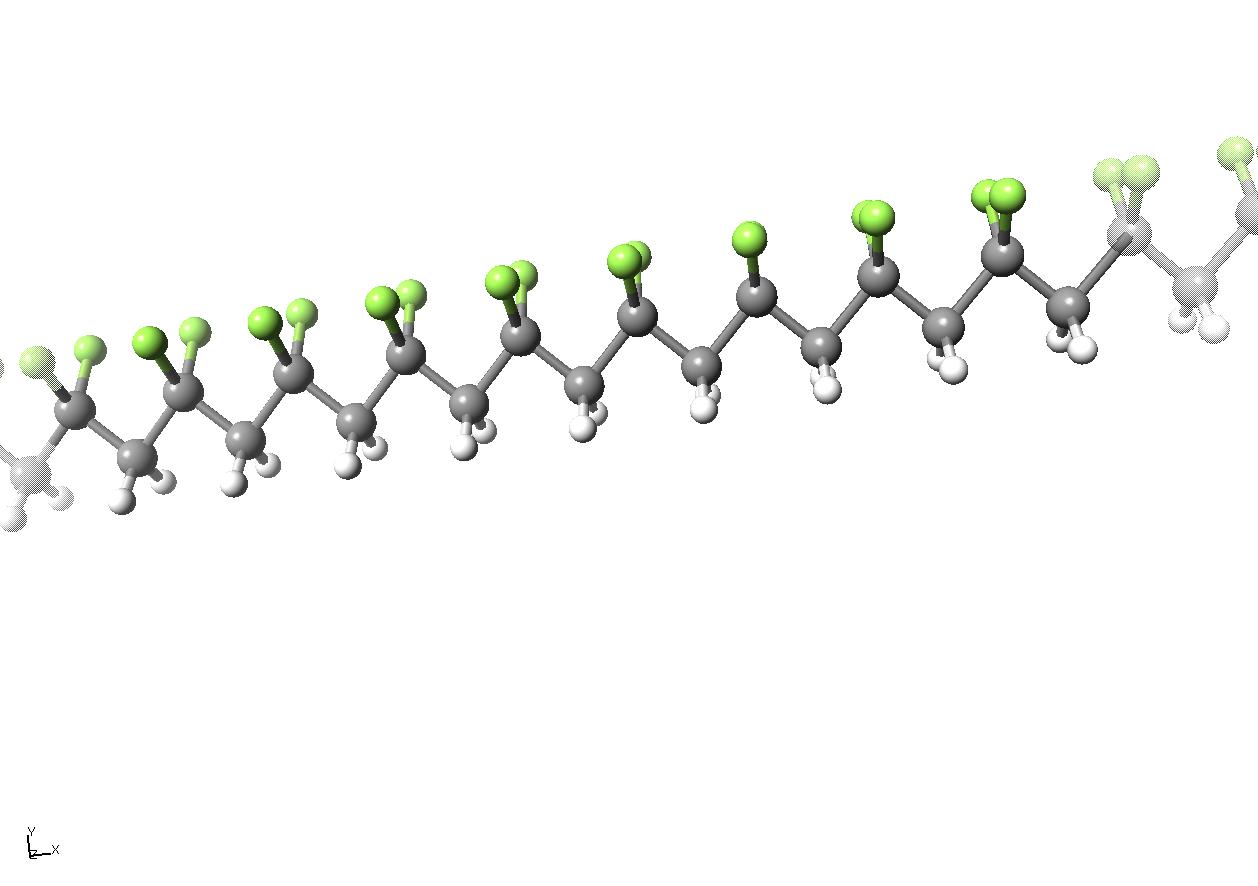 図球棒モデル(ボールスティックモデル)の3DCGを使ったPVDFの分子模型
(©M.Akama)
図球棒モデル(ボールスティックモデル)の3DCGを使ったPVDFの分子模型
(©M.Akama)
SCIGRESS
実習室のWindows PCにログオン後
[スタート] → [SCIGRESS 2.8.1] → [Workspace]
計算例)
フェリシアニド([Fe(CN)6]3-)錯体の分子軌道計算
同時使用ライセンス(フローティングライセンス)
80プログラムの同時起動が可能
分子軌道計算では初期配座が重要
ChemSpider:http://www.chemspider.com/
alpha-Cyclodextrinで検索する。3D表示する。mol形式でダウンロードする。
サイグレス
で開く。
シクロデキストリンの3D表示
構造最適化を行う。MOに関してはライセンスの上限があるので先着順です!
構造最適化が終わったらHOMOとLUMOの波動関数を計算する。
アセトニトリルのHOMOとLUMO
エネルギー化学への応用
量子力学計算を
エネルギー化学に応用できます。
波動関数のエネルギー準位と酸化還元電位の関係を調べることができます。
酸化還元電位の算出
HOMO軌道とLUMO軌道のエネルギー順位をSCIGRESS(PM6 geometry with PM6 wavefunction※1)を用いて計算すると下図のようになった.
Fe3+のLUMO軌道のエネルギー準位は,-30.23eVとなり,Fe2+のエネルギー準位は-28.68eVとなった.
さらに,水素分子(H2)のHOMO軌道のエネルギー準位は,-15.04eVとなった.
水素イオン(H+)のLUMO軌道のエネルギー準位をSCIGRESSで計算すると,「No surface was found for molecular orbital 1 since it does not contain a value as large as 0.060.」とエラーが表示されLUMO軌道のエネルギー準位を計算できない.
Waiting for resources to become available...
Keywords used in this job are as follows:
PM6 NOMM NOXYZ XYZ NODIIS GRAPH T=10D CHARGE=1
MO-GManager 1.1.1.0 Apr 2 2018 21:50:58
MO-GComputeEngine 2.9.0 Apr 2 2018 21:50:15
Final heat of formation = 311.4630 kcal/mol
Calculation done.
TabulatorManager 2.9.0 Apr 2 2018 21:45:33
TabulatorComputeEngine 2.9.0 Apr 2 2018 21:44:57
3 3 3 (529169.25,529169.25,529169.25) Angstrom (-529169.25,-529169.25,-529169.25) Angstrom Molecular orbital values: 0.060, -0.060 Using molecular orbitals 1 - 1
No surface was found for molecular orbital 1 since it
does not contain a value as large as 0.060.
Computation time: 00:00:00.388
酸化還元電位の計算
ギブスの自由エネルギーの計算はどうやるの?
Gaussianでギブスの自由エネルギーを求める方法は下記のキーワード検索すると見つかることがあります.
検索キーワード:gibbs free energy gaussian Google
文献などのExampleを探しましょう.基本は,物理化学
新型コロナウイルスの影響による在宅課題
分子軌道計算ソフトを使ったα-シクロデキストリンのHOMO-LUMOエネルギーの計算
- Winmostarのインストール
- Winmostarのインストールをしてください.学生の方は学生版が利用できます.あとは,ウェブページの指示に従ってインストールしてください.
本学の電子メールの使い方がわからない方は,「学生向け電子メールの使い方」をご覧ください.
- α-シクロデキストリンの3次元のmolファイルをダウンロード
- Chemspiderを使ってα-シクロデキストリン(alpha-cyclodextrin)の3次元のmolファイルをダウンロードしてください.
- Winmostarの起動
- Winmostarを起動して,molファイルを開いてください.
- 構造最適化
- 構造最適化を行います.メニューバーの「半経験QM] -> [MOPAC] -> 「MOP7W70の実行」をクリックしてください.
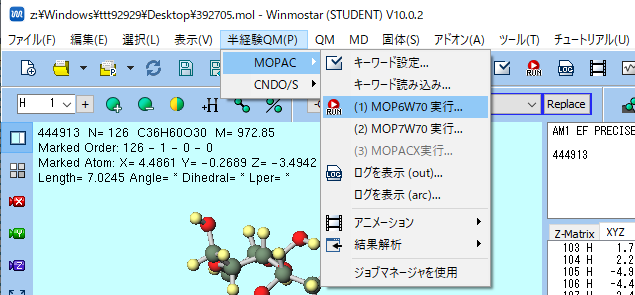 MOPAC Setupのダイアログウィンドウが起動したら,OKをクリックしてください.
ファイル名の保存ダイアログウィンドが立ち上がった[保存]をクリックしてください.なお,ダイアログウィンドウは,起動しないこともあります.
メモ帳が軌道したら,最適化完了です.
MOPAC Setupのダイアログウィンドウが起動したら,OKをクリックしてください.
ファイル名の保存ダイアログウィンドが立ち上がった[保存]をクリックしてください.なお,ダイアログウィンドウは,起動しないこともあります.
メモ帳が軌道したら,最適化完了です.
- エネルギー計算
- エネルギー計算を行います.メニューバーの「半経験QM] -> [CNDO/S] -> 「実行」をクリックしてください.
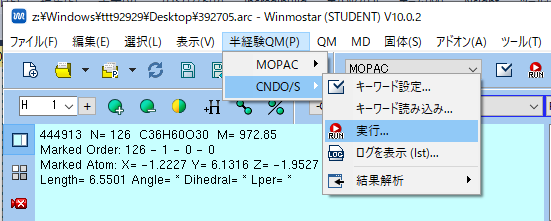 CNDO/S Setupのダイアログウィンドウが起動したら,OKをクリックしてください.
ファイル名の保存ダイアログウィンドが立ち上がった[保存]をクリックしてください.なお,ダイアログウィンドウは,起動しないこともあります.
CNDO/S UV-Vis-Spectrumなど3つのダイアログウィンドウが起動したエネルギー計算完了です.
CNDO/S Setupのダイアログウィンドウが起動したら,OKをクリックしてください.
ファイル名の保存ダイアログウィンドが立ち上がった[保存]をクリックしてください.なお,ダイアログウィンドウは,起動しないこともあります.
CNDO/S UV-Vis-Spectrumなど3つのダイアログウィンドウが起動したエネルギー計算完了です.
-
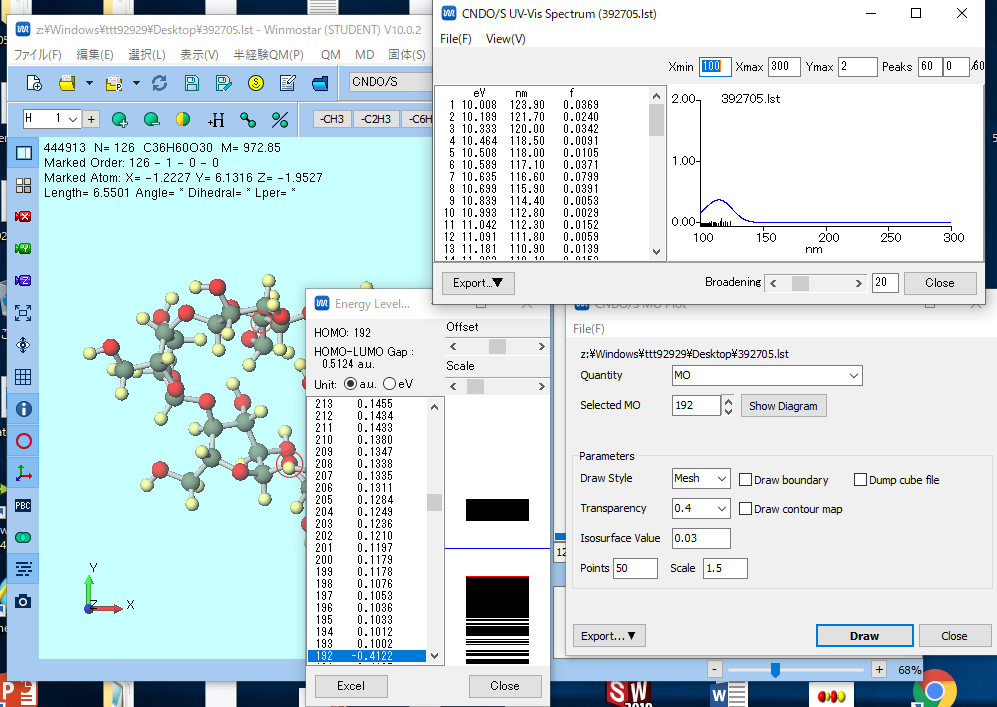
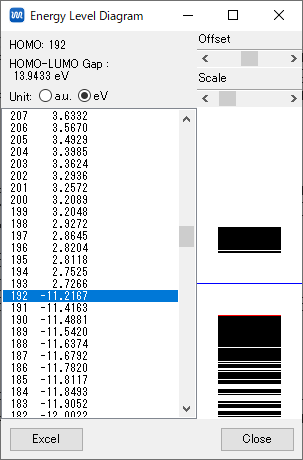
HOMO LUMOエネルギーの表示
- Energy Level… ダイアログウィンドウを見てください.HOMO:の後の数字と同じUnitの値がHOMO軌道のエネルギーになります.その値を読みとりウェブクラスに提出してください.
また,HOMO:のUnitの値に1を加えた値がLUMO軌道となります.LUMO軌道のエネルギーの値をウェブクラスに提出してください.
 🏠
🔋C1 Laboratory
🏠
🔋C1 Laboratory
 化学式ワープロと分子軌道計算~量子力学もコンピュータで解こう~
化学式ワープロと分子軌道計算~量子力学もコンピュータで解こう~
