.jpg "図2.2 原子のエネルギー準位と固体のバンド及びバンドギャップ")
東北大学選鉱精錬研究所教授 小沢昭弥
編集同志社大学工学部教授 山下正通
東京理科大学工学部教授 小浦延幸
神奈川大学工学部教授 佐藤祐一
国際化時代にふさわしい英語力を身につけると共に、学生にやる気をおこさせる教科書を出版してみたいと考えたのは約15年前です。それは私がUCC社の代表としてアメリカ化学会の夏講座でアメリカの大学の若い助教授クラスの先生方に、工業の第一線にある研究者の希望として大学でこんな教育をしてほしい、ということを示すために電池の講義*を3日間行なった時のことである。
(A. Kozawa and R.A. Powers, J. Chem. Ed. 49, 587(1972))
要するにテキストが非専門家により書かれていて、すべての問題がすでに解決されてしまっているかのような印象を学生に与えてしまうことが教育上よくないのです。専門の研究者からみると、「こんなにはっきりと言いきってよいのかな?」という部分が今のテキストには多すぎるのです。学生にやる気を起こさせるには、「定説や例外も多く必ずしも正しくない」とか「これから社会環境問題についてこんなに沢山の研究テーマがある」とか、今後化学者として開発すべき理論や解決すべき問題が沢山あることを示すことにより学生にやる気を起こしてもらえるのです。こんなことを考えてこの本の編集にあたったのです。
理工英語教育については特別の理工英語コースを設定することなく、専門コースの中で教育してゆくよい方法を確立したいと念願し、理工英語のテープを配布する学会を作り色々と20年以上努力してきました。(化学と工業, 32, 934(1979))。
このテキストは専門家による各分野を執筆して戴くことと専門教科の英文や用語のテープ付のテキストを作るという2つの目標をたてて、執筆者にお願いしましたところ、多数の専門家の先生方のご参加を戴きここにテキストとして発刊できることになりました。執筆者の先生方に厚く心から感謝を申し上げるものであります。
なお、本書の編集にあたり、熱意をもって多大の時間をおさき下さった編集者の山下正通先生、小浦延幸先生、佐藤祐一先生に心からお礼を申し上げます。
電気化学は、物理化学の基礎から応用までの主要な部分を占めると同時に、今や化学の広い分野でその原理が適用され、また重要な各種測定手段としても活用されている。すでに完成された学問として、一時衰退したかにみえたこの分野が、いまやふたたび新たな飛躍を求めて、活況を呈しているのである。たまたま、本書の執筆、編纂中に”常温核融合”という未だ決着のついていないセンセーショナルなできごともあった。このような状況下で、電気化学がいかに魅力的な学問分野であるかを学生諸君に伝えたいという意図のもとに、大学用テキストとして、テクニカルタームとその応用論文(Yeager教授の学会賞受賞論文)の発音テープを併用した<新形式の電気化学テキスト>を編纂することを考えた。本書の構成には若干の苦心をはらったが、幸い各章の執筆者には、その分野の第一線でご活躍中の方々のご快諾を得、完成させることができた。
第1章では電気化学発展の歴史と概念が述べられ、第2章では以後の各章が理解できるよう電気化学の基礎事項が述べられている。第3章以降は多少強引かとも思われる分け方で、分野別に分けその基礎事項から未だ進展中の最先端までの内容を執筆して戴いた。また、各章ごとにテクニカルタームを挙げ、その発音をテープにおさめた。近い将来、国際学会で、あるいは業務上、外国の人たちと接触される場合の動機付けとなれば幸いである。なお、各章間の連携と統一に心がけたが、欠けているところも多いと思われる。それらは編者の責任であり、御許し戴きたい。
最後にご多忙中、快く執筆を引き受けられ、長期間にわたって辛抱強くわれわれとつきあって下さった先生方、また出版を引き受けて下さった新星社永井孝英氏にお礼申し上げます。
1990年9月
編集者 山下正通
小浦延幸
佐藤祐一
2021年2月9日に、内田勇先生はご逝去されました。
イタリアの解剖学者Lugi Galvani(1737-1798)は、蛙の解剖に端を発した二つの異種金属を接触させたときに流れる電流を動物電気と称した(1979)。 この現象は直ちに同国のAlessandoro Count Volta(1745-1827)により追試され、ボルタの電堆として実証された(1800年3月)。 Galvaniの業績をたたえてこの種の電池を ガルバニ電池と呼んでいる。
一方、Galvaniの発見した動物電気、すなわちメス(解剖刀)の接触による蛙の足の伸縮運動の現象は、神経細胞の興奮の仕組みを解明する切っ掛けを与えた。この動物電気は異種金属の接触に依り生じた刺激をイオンの移動による電気化学的化つまりの細胞膜電位に変換するもので、これは生体受容器(BiologicalTransducer)あるいは神経トランスミッター(Neurotransmitter)の研究に発展し今は、現在のメディカル・エレクトロニクス(Medical Electronics)、バイオ・エレクトロニクス(Bioelectronics)、モレキュラ・エレクトロニクス (MolecularElectronics)およびエレクトロオルガニック・ケミストリー(ElectroorganicChemistry)などの分野開拓への一里塚となった。なお、光の電気化学への参入は、Alexandre Edmond Becquerel(1820-1891)による溶液中の電極の光起電力の発見(1839)に端を発している。その後、W.G.AdamsおよびR. E. Day により固体素子セレンの光起電力効果(Photovoltaic Effect)が電極の半導体的性質によるものであり、液中物質の光化学変化による光ガルバニ効果 (Photogalvanic Effect)と区別されるようになった。さらに、水電解により生成した水素と酸素の系について、Sir William R. Grove(1811-1896)は水素・酸素燃料電池を考案した(1839)。この電池のエネルギー変換効率について Friedrich Wilhelm Ostwald (1853-1932)はカルノーエンジン(Carnot Engine) の制約を受けないことを示唆した(1894)。なお、この電池の起電力はOstwaldの弟子であるHermann Walter Nernst(1864-1941)によって誘導された可逆電極電位の理論式であるネルンスト式(1900)から算出できる。最初、彼は金属がそのイオンを溶液中に放出する傾向を尺度として電離溶圧という概念を導入し、溶液中から金属イオンが電極面に析出する傾向は溶液の浸透圧に比例するものと考えた(1889)。しかし、R. A. Lehfeldtは電離溶圧の物理的意味はないと看破している(1899)。その後、Nernst は Gilbert Newton Lewis(1875-1946)の提唱した活量の概念(1907)をその式に導入した。例えば、Edward Weston(1850-1936)が開発したウエストン型標準電池[Cd(12.5%amalgam) | CdSor(s) | CdSO(aq,satd) || HgSO(s) |H3(1)]の25 ℃ での電位はネルンスト式を適用すると1.0180Vとなる。なお、P.Henderson は液間電位の理論式を提唱した(1907)。さらにOstwald の教え子である Frederick G. Donnan(1870-1956)は半透膜を介しての高分子イオンの膜平衡電位をネルンスト式を適用して求め、その現象をドナン平衡と称している(1911)。その他、W. R. Hainsworth および D. A. MacInnes による可逆性電圧に対する圧力の影(1924)など多くの研究がある。また、Lewis の教え子である Wendell M.Latimer(1893-1955)は水溶液系でのイオン種の標準酸化還元電位を熱力学的立場から算出して、その電位図をラティマー線図と呼称している(1952)、すでにJ. J.Hermans はKCI塩極法による可逆電圧の測定値は数mV の精度までは信頼できるが、それ以上の精度の測定は困難であることを指摘している(1939)。
この間、Svante A. Arthenius(1859-1927)のイオン解離説が提唱され(1883)、Ostwald によって支持された。水電解に関連して、Paul Erman(1764-1851)は電界質の濃度を増やすと電流が増加することからモル導電率の濃度依存性を予測した(1801)。これはFriedrich Wilhelm Kohlrausch(1840-1910)により実証され、Kohlrausch のイオン独立移動則(1876)および強電解質系でのコールラウシュの平方根町(1900)として確立された。その後Peter J. W. Debye(1884-1966)および彼の教え子の Erich Hückel(1896-1980)によるイオン活量係数に関するデバイ・ヒュッケルの理論(1923)、非平衡熱力学で有名な Lars Onsager(1903-1976)によるオンサーガーの式(1927/1932)などが導入されると共に、N. Bjerrum のイオン会合の概念(1926)、M. Wienの導電率に対する高電場の効果(1927)、P.Debye および H.Falkenhagen の導電率に対する高周波電場の効果(1928)などが見い出された。 すでに、電極との界面における電荷に対する関心が、Hermann LudwigFerdinand von Helmholtz(1821-1894)によって示され、いわゆるヘルムホルツの固定二重層モデルが提案されていた(1879).この分野の研究は前述のアレニウスのイオン解離説(1887)の出現により、Louis Georges Gouy(1854-1926)およびDavicLeonald Chapman(1869-1958)による拡散電気二重層モデル(1909/1913)、OttoStern(1888-1969)によるいわゆるシュテルンの固定・拡散二重層モデル(1924)へと発展していった。さらに、David Caldwell Grahame(1912-1958)による電気二重層での特異吸着現象(1947)と、その電気二重層モデルが M. A.V.Devanathan、」O'M. Bockris、Klaus Mullerによって提案された(1963)。 一方、Julius Tafel(1862-1918)は水素過電圧と電流密度との関係を速度論的過程として捉えたターフェル式を提唱した(1905)。また、Boltzman統計および速度論平衡条件に基づく John A. V. Butler(1899-1977)による可逆電極電位の理論(19231924)、ついで、Nernst の教え子であるMax Volmer(1885-1965)の実証により電通一過電圧曲線の一般式である Butler-Volmer 式が確立した(1930) その後、Aleksands N. Frumkin (1895-1976)は電極反応速度論の立場から、電荷移動過程(または活性化過程)に及ぼす電気二重層の影響を論じた(1933)。さらに、混成電位の速度論的な考え方が A. N. Frumkin、Carl Wagner(1901-1977)らによって提唱(1932/1938)されるに及んで、いわゆる、"The great Nernstian hiatus" を克服した。 その間、志方益三(1895-1964)と一緒にポーラログラフィーを発明(1924)したJaroslav Heyrovsky(1890-1967)は、彼の教え子である Dionyz Ilković(1907-1980)と共に拡散電流についてのヘイロウスキーイルコヴィッチ式を誘導した(1935)。 その後、国際的にも電気化学の見直しが盛んに行われ、エネルギー変換・貯蔵、環境浄化・検知、腐食・防食、新素材開発、など境界領域の分野の研究にも関心が向けられるようになってきた。
さて、電気化学とは何かという命題に対して、定説はいまだ見当たらないのが現状である。 したがって、従来の定説にこだわることなく、新しい分野を切り開いて行けばよいことになる。
もし、<電気化学>という言葉が古典的で、これまでのヒエルラルキー(Hierarchie)を忠実に守る学問という先入観があるとすれば、 競争原理の中でとられる模倣Emulationも無徒ではなかろう。それは結局<大師は弘法に>の言葉が意味するように、 国際的視野に立脚して、これら分野の成果を集約して表現すべきであろう。 例えば、将来の展望を踏まえて<電気化学>とは、『イオン、電子、粒子、導体、半導体、誘電体間の界面および本体における荷電粒子の存在と移動を制御する科学と技術』 と定義しておこう。
電気化学の特徴でもある学際ないしは境界領域のこの分野の進展を目指して学ぶことにしよう。 例えば、国際電気化学会の専門部会 (①基礎界面電気化学Fundamental Interfacial Electrochemistry、 ②電極と電解質材料Electrode and Electrolyte Materials、 ③分析電気化学Analytical Electrochemistry、 ④分子電気化学Molecular Electrochemistry、 ⑤電気化学的エネルギー変換Electrochemical Energy Conversion、 ⑥腐食・電析と表面処理Corrosion, Electrodeposition and Surface Treatment、 ⑦工業電気化学と電気化学工学Industrial Electrochemistry and Electrochemical Engineering、および ⑧生物電気化学Bioelectrochemistry) を横観してみれば、いかに電気化学の分野が多岐にわたっているかが伺える。 さらに、所属会員数の最も多い米国電気化学会では、12部門(Division ①電池Battery、 ②腐食Corrision、 ③誘電体科学と技術Dielectric Scionce and Technology、 ④電析Electrodeposition、 ⑤エレクトロニクスElectronics、 ⑥エネルギー工学Energy Technology、 ⑦高温材料High Temperrature Materials、 ⑧工業電解と電気化学工学Industrial Electrolysis and Electrochemical Engineering、 ⑨蛍光と表示材料Luminescence and Display Materials Group、 ⑩有機生物電気化学Organic and Biological Electrochemistry、 ⑪物理電気化学Physical Electrochemistry、 ⑫センサーSensor Group) に分類して活動を行っている。特に、個体イオニクス(Solid State Ionics)関係の分野の進展が著しいのが現状である。
かように、電気化学の領域は、純正自然科学(理学)的な場と応用自然科学(技術・工学)的な場との両面にわたっており、共通した基盤の上に成り立っている。
◇電気化学系の最小単位は、ひとつの電子伝導体Mとひとつのイオン伝導体Sの接触によって構成される。……
電気化学系の最小単位は、一つの電子伝導体Mと一つのイオン伝導体Sの接触によって構成されます 1 ) 。 最小単位をセルと言います。電池や電解槽の最小単位です。
電解質は、電子絶縁体です。 電池で、電子絶縁破壊が起きると、電気分解です。 コンデンサでは、電子絶縁体は、誘電体です。
固体内の電気伝導機構は、電荷を運ぶキャリアーと呼ばれるものの種類によって2つに分けられる。キャリアー(carrier)が電子(electron) あるいは本来あるべき電子が抜けた状態(正孔(positive hole)と呼ばれる)の場合を電子伝導、キャリアーがイオンの場合をイオン伝導と呼んでいる。電子伝導の場合、伝導性の論議には、次に説明するバンド理論 (band theory)を使うとわかり易い。
原子あるいは分子が孤立して存在する場合、それらに所属する電子は、原子軌道あるいは、分子軌道の一番低いエネルギー準位から、パウリの禁則にしたがって互いにスピンを逆にした電子対となって順にうまっていく(図2.2)。 エネルギーのい方には、空の軌道に相当するエネルギー準位が存在する。原子、分子が集合して固体を形成すると、同じく図2.2に示すように、各エネルギー準位は相互作用の大きさによって、大小のエネルギー幅をもったバンド (band,帯)として固体全体に広がって存在する。電子が満ちたバンドを充満帯(filled band)、空のバンドを空市(empty band)と呼び、それらのバンドの間のエネルギーには電子は存在できないので禁制帯 (forbidden band)と呼んでいる。エネルギーの一番低い空帯を特に伝導帯 (conduction band)と呼び、エネルギーの一番高い充満帯との間の禁制帯の傷をバンドギャップ(band gap) と呼んで、Eで表わす。バンドが空の時はもうん、電子がいっぱいに詰まってしまうとキャリアーが存在しないので電気伝は示さない(いっぱいに詰まった状態では、電子はキャリアーになり得ない)。固体全体に広がっているバンドにおいて、空の伝導体に電子が入ると、その電子はキャリアーとなって固体全体を自由に移動できるので、自由電子 (free electron)と呼ぶ。金属の場合、電子の入っている一番エネルギーの高いバンドは電子がいっぱいになっていない状態なので、キャリアーである自由電子が伝導体にたくさんあることになり、導電性が高い。絶縁体(insulater)も真性半導体(intrinsic semiconductor)も、バンドギャップの大きさが違うだけで、絶対零度ではバンドはすべて充満帯と空帯だけでできているので、キャリアーは存在せず、電導性は示さない。しかし、バンドギャップが小さい真性半導体は、温度を上げると、図2.3(a)に示すように、充満帯にある電子が熱励起されて、伝導帯に飛び上がり、自由電子となる。また、充満帯には電子が抜けたので、正孔ができる。どちらもキャリアーとなるので、真性半導体は温度を上げると導電性がでてくる。熱励起されて伝導帯に上がる電子の数は、温度とともに指数関数的に増えるので、導電率も指数関数的に大きくなる。絶縁体はバンドギャップが大きいので、室温程度では電子を熱励起できなくて、導電率は非常に小さい。

上式は、単極電位に関するNernst式と呼ばれる。
ここでは、ある有限な大きさの電流が電極系に流れ、電極ー電解液界面で起こる。 反応が不可逆となる条件下での、電極反応速度と電極電位との関係について述べる。
電解に際し、電極系に流れた電気量(q)と電極反応によって生成する物質のモル 数(n)との間の定量関係がFaradayにより、1833年に提案された。その Faradayの法則(Faraday's low)は次式で表示される。
q=nF (2.84)
ここで、FはFaraday定数と呼ばれ、その値は9.6485×104C(クーロン)・mol-1であ ることが実験的に求められた。この関係は、電解反応に限らず、すべての電極反応 に適用される。また、複数の電極反応が、並列又は連続して起こる場合も、各電 極反応ごとに使われる電気量と生成物の間には、(2.84)の関係が成り立つ。 さてここで、次の反応を考えてみよう。

|dq|=nF|ν|dt (2.87)
電流密度をJで表すと、
νa=ν++ν- (2.89)
Ja=J+-J- (2.90)
J+は電極上での酸化反応に伴って電解液から電極に向かって流れ、J-は、電極上 での還元反応に伴って電解液から電極に向かって流れる電流である。したがって、 両者の差が正味の反応速度、すなわち正味の電流ということになる。一方、2.2節で みた可逆電極電位Erevは、電極反応の酸化および還元両方向の速度又は電流密度 の絶対値が等しい、いわゆる動的平衡状態において示す電位である。ν+=|ν-|≡ν0 (2.91)
J+=|J-|≡J0=nFν0 (2.92)
ここでν0は交換反応速度と呼ばれ、これに対するJ0は交換電流密度(exchange current density)と呼ばれる。電池または電解系において、外部負荷または外部電源に接続すると、系に電流J を流すことができる。J>0の場合には、電極電位(E)が可逆電極電位(Erev)に 比べ貴にシフトし、J<0の場合はその逆にシフトする。両電位の差、
η=E-Erev (2.93)
は電極反応の不可逆性を示す尺度で、過電圧(overpotential)と呼ばれる。過電圧 をかけて電位を貴にシフトさせると部分アノード電流J+と部分カソード電流J-が 流れることになるが、J+≫J-で、J+-J-=Jの不可逆分だけ正味のアノード電流が 流れる(図2.19参照)。ここに示した過電圧は大別して、i)電荷移動反応に関する 活性化過電圧(ηa)と、ii)物質移動家庭に関係する濃度過電圧(ηc)とからな る。前者には、電子授受過程とそれの先行または後続の化学反応過程が関与する。次節以降で、これらの反応過程の過電圧及び電流密度の関係について述べる。

今、次に示す簡単な電極反応を考える。
X + ne ⇔ Y
その電流密度は次式であらわされる。
図1.(2.114)に記載。*以降の式をレポート用紙にまとめる。
ここで、k₊及びk₋は式(2.96)及び式(2.97)に用いたものと同じである。Cx(t)、Cy(t)はそれぞれ電解開始後、時間tにおけるX、Yの電極表面濃度を示す。J=η=0 の場合は常に Ca(t)=Cx°、Cx(t)=Cx° とみなせる。ただし、Cx°、Cy° は容液内部でのX、Yの濃度である。しかし、J≠0(n ≠ 0)の場合は、t = 0の場合を除いてこの関係は成り立たず、定常状態に達するまで表面濃度は時間と共に変化する。すなわち、Jまたはηの少なくとも一方は、時間の関数となって変化する。
物質の拡散が線拡散の場合、詳細は他書に譲るが、Fick の第二法則を解いて、結局次式が得られる。

(2) 定常物質拡散

もし、反応に関わる各過程中、物質移動が著しく遅い場合、その反応電流は反応物の移動速度に比例する。移動が拡散のみによる時、その電流を拡散支配電流(diffusion controlled current)と呼ぶ。 電極面における濃度勾配が、電極面と溶液層内部との濃度差(C0xーCx)を拡散層の厚さδxで割ったものとみなせるNernstの拡散層モデルが満足される場合、イオンが電極面に到達する速さ、すなわちイオンの拡散速度は濃度勾配に比例する。 その比例定数Dxは拡散係数(diffusion coefficient)と呼ばれ、それと濃度勾配の積Dx(C0xーCx)/δxは拡散速度を表し、反応電子数nとファラデー定数Fをかければ、次式で示す拡散電流(diffusion current)が求まる。
J=nFD_x (C^0_x-C_x)/δ_x=nFk_x (C^0_x-C_x) (2.117)
ただし、k_xは拡散過程の速度定数ともいえる量である。
したがって、電極面濃度C_X、C_Yは次式で与えられる。
C_X=C^0_x-J/nFk_X
C_Y=C^0_Y+J/nFk_Y
上式を基本式(2.103)に代入し、次式が得られる。
J/J_0=(1-J/nFk_XC^0_x)exp((α+nF)η/RT)-(1+J/nFk_YC^0_Y)exp(-(α-nF)η/RT) (2.118)
nFη/RT>>1の極限では上と同様に近似的に、
J/nFk_XC^0_x=1
となり、したがって、
J=nFk_XC^0_x≡J_l+ (2.119)
となる。同様にして、-nFη/RT>>1の極限では、
J=-nFk_YC^0_Y≡J_l- (2.120)
となる。ここで、 J_l+、J_l-は、アノードおよびカソード反応における拡散支配の限界電流密度(limiting current density)である。この値を用いて(2.118)式を書き直すと、
J/J_0=(1-J/J_l+)exp((α+nF)η/RT)-(1-J/J_l-)exp(-(α-nF)η/RT) (2.121)
が得られる。電流―過電圧曲線が交換電流、限界電流、移動係数に依存することがわかる。J_0、J_l、のJ-η曲線への影響について、一例を図2.20に示した。

p.62-64 第3章電池とエネルギー
化学電池を、酸化還元反応に伴う自由エネルギー変化を電気エネルギーとして取 り出す仕組みを持つ装置、すなわち化学エネルギー変換(energy conversion)デバ イスと呼ぶことにしよう。これは太陽電池などの物理現象を利用する物理電池とは 異なる。電池は本来、独立電源としての機能を有し、コンデンサーのような従属的 受動的な電気エネルギーの~時的な溜池とは異なる。また、最近の電池は益々小 型·軽量化され腕時計,カメラメモリーのバックアップ等では、数年から10年間 もの寿命が保障されている。まさに、コードレス化エレクトロニクス時代を支える 電源であり、能動的なデバイスである。
本章ではその化学エネルギー変換の原理と仕組み、電池内酸化還元物質の化学、 変換過屋のキネティクス、代表的な実用電池と実用化の期待されている新型電池に ついて学ぶ。
常温等圧化における化学反応の最大エネルギー変化は ギブスの自由エネルギー変化(ΔG)に等しい。ある化学反応が完結するまでに放出されるであろうこのΔG (Jまたは、kJ/molの単位をもつ)がすべて 電気エネルギー (クーロンボルト、 CVの単位をもつ)に変換されたとすると、
Fはファラデー定数(電子の電荷(C)×アボガドロ数(N))。ここで、電子数(n) と電圧(E)の意味について以下のような電池反応を想定して検討してみよう。 電池反応は必ず2つの半反応(half action)の組合せからなる酸化還元反応で あり、半反応1と半反応2を次のように表わして、為とnの最小公倍数を用いて電 子を含まない反応式を作れば、これが電池反応式(3.Δ)に他ならない。 (3.2) Ox1+ne1=Red1 Ox2+ne2=Red2 n2×(3.2)-n1×(3.3)2nOx1+nRed2=n2Red1+n1Ox2 ΔGは(3.Δ)式のそれであり、nは(3.Δ)式に関してやり取りされる電子の数であ るから、ここではn=n1n2である。また、E cellは(3.2)、(3.3)式の平衡電位(ネルン スト電位)の差、E1=E2-E1になる。ここで、nの方にはn1,n2が含まれるの に、E。の方にはそれらが含まれないのは不思議に思うかも知れない。これは、 と(3.3)の間の引算をΔGに注目して行ってみるとよく理解できる。
(3.2)×n2 n2Ox1+n1n2e=n2Red1 -ΔG1=n1n2FE1 (3.3)×n1 n2Ox1+n1n2e=n2Red2 -ΔG2=n1n2FE2 -ΔG=nfE cell =ΔG1-(-ΔG2)=n1n2F(E1-E2) (3.5)このように電位は示強因子であるから、量論的な数には無関係である。 E cellを(3.Δ)式の電池反応で示される電池の起電力という。これらを標準状態で扱えば、E cellは標準電極電位の差(E2-E1)で与えられる(図3.1参照)。 上の説明は、(3.1)式が与えられるとして、電池反応を想定してnおよびE cellの後付けしたものであるが、(3.2)、(3.3)式の半反応から出発して(3.1)式を導くのが本来のやり方である。他の成書を参考にされたい。(3.1)式は酸化還元反応のΔGが直接電気エネルギーに変換されることを意味しており、熱エネルキギーを経由しない(燃焼反応も、燃料の酸化、酸素の還元からなる酸化還元反応である)高効率なエネルギー変換の可能性を示している。 電池の理論エネルギー変換効率(theoretical energy conversion efficiency)は、 熱エネルギー(ΔH)をベースにとれば、 εth=ΔG/ΔH (3.6) Eth=ΔG/ΔH すなわち、熱として利用できるのはΔHであるが、電気化学的にはΔGがそっくりそのまま電気エネルギーに変換できると言うわけである。ΔG/ΔHは多くの場合ほとんど1に近い値で、電気化学的エネルギー変換の効率が100%に近いというせっかちな結論がここから出てきてしまう。周知のように、ΔGはあくまでも熱カ学的量であって、定義の中には「可逆変化に際して」という注釈が入っている。したがって、ΔGに対応する(3.1)式のE cellに対しては、「電流ゼロにおける電位差の極限値を持ってE cellと定義する」という電気化学的注釈が入る。現実には、E cellとは電流を流さない開路状態の電圧であって、電流を出力しているときの電池の出力電圧(E)と同じではない。実際の電池効率(s)は電圧効率(E/E cell)と電流効率を含む。電流効率は電池活物質の利用効率で置き換えることができるから、 Εac=(ΔG/AH)(E/E)(Q/Qo) (3.7) QとQoとは、それぞれ現実に電池から取り出された電気量と電池内に有する活物質(active material)の理論電気量である。また、Eは電流(I)の関数であり、これらについては後段で触れる。電池のエネルギー変換の効率は、したがって、出力が電流ゼロに近づくほど、理論効率に近づくと言い直すべきである。ところが、これから述べる電池はすべて電流を出力する(出力W=IE)ことを目的とする実用電池であって、電流を取り出すことを目的としないタイプの電池(イオン電極など電位差を読み取る形式のセンサー類も基本的には電池の構成をしている)とは区別される。実用電池のおもしろさと難しさとは、この(3.7)式中の(E/E cell)と(Q/Qo)をいかに1に近づけるかにある。

古典的な電池にダニエル電池(Daniel cell)がある。これを用いて、上の問題をもう一度復習してみよう。
ダニエル電池の電池反応は、
Cu 2+ + Zn → Cu + Zn2+
半反応と電極電位は、
Cu 2+ + 2e=Cu Ee(Cu 2+/Cu)=0.337+(RT/2F)ln aCu2+ (3.8)
-)Zn 2+ + 2e=Zn Ee(Zn 2+/Zn)=-0.763+(RT/2F)ln aZn2+(3.9)
Cu 2+ + Zn = Cu + Zn2+ Ecell=1.100V (3.10)
それぞれの活量は等しい(aCu2+=aZn2+)ものとした。Eo(Cu2+/Cu)>Eo(Zn2+/Zn)であるから、電池の表記はZn|Zn2+¦Cu2+|Cuである。
ダニエル電池の放電の様子を図3.2に示す。正極反応では、Cu2+は外部回路から電子を受け取ってCuとなり、負極反応ではZnがZn2+として溶解し、外部回路へ電子を放出している。すなわち、 電池反応を支えるそれぞれの半反応の方向は逆である 電気化学では、電子を受け取る反応をカソード反応、電子を放出する反応をアノード反応といい、反応の進行する方向を区別している。したがって、 正極反応はカソード反応であり、負極反応はアノード反応と覚えていただきたい。日本語の感覚では、カソード(cathode )=陰極、アノード(anode )=陽極という図式があって、このイメージで極性を連想す ると極性に関する混乱が起きてしまう。これからは、陰極、陽極という言葉は使わないことにしよう。電気化学は溶液側から電極を眺めているのであるから、カソードとは電子を受け取る 場所(electron reservoir ) で、アノードは電子を外部回路へ放出する場所(electron sink )と考える。
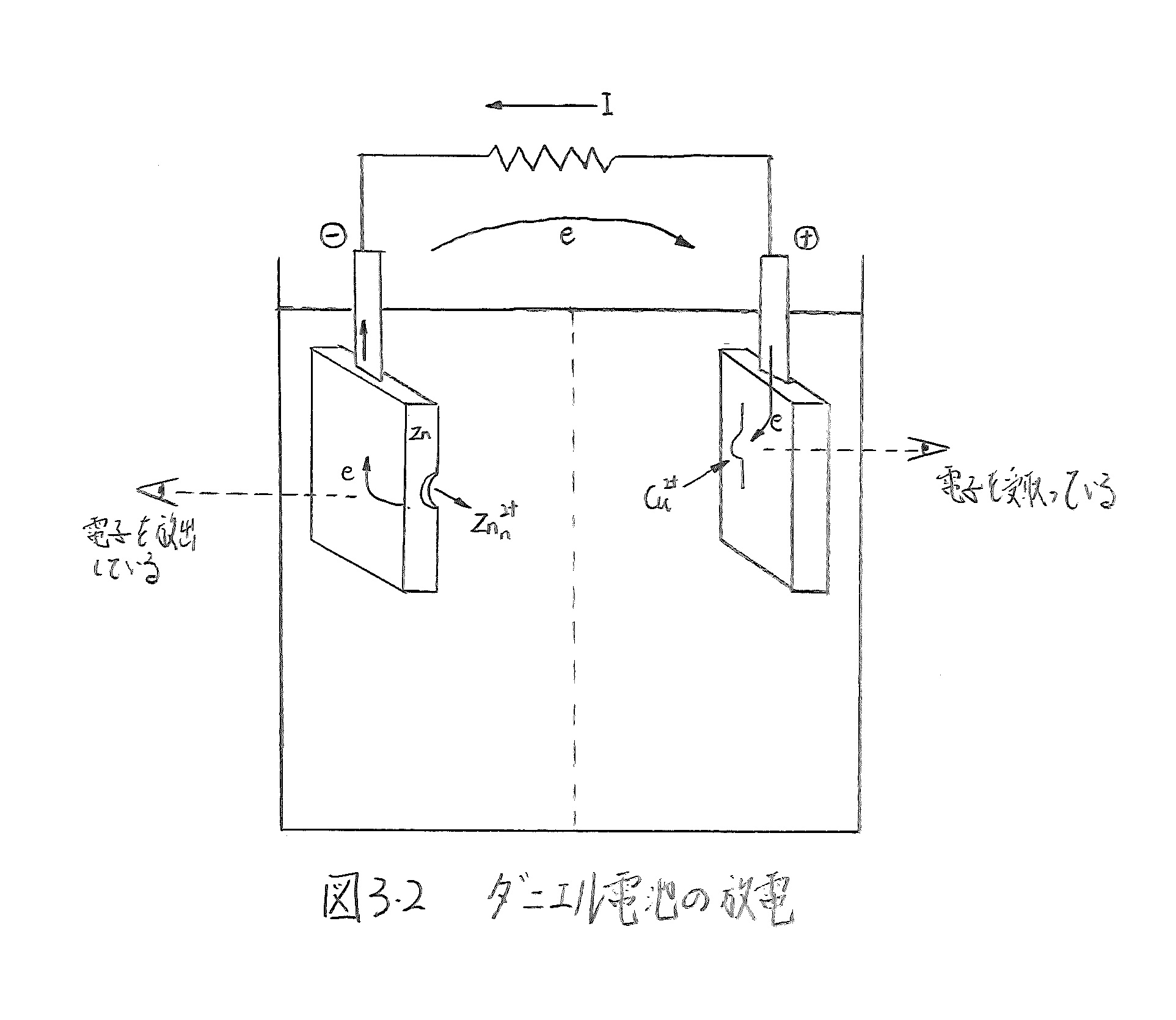
極性に関するもう一つの混乱は、いわゆる電極電位(EoまたはEeで表記し、標準水素電極を基準としている)と電池の起電力(Ecellで表記し、 Ecell=E1-E2)との定義上の違いから生ずる。電極電位は標準水素電極を必ず左側において構成した電池 の起電力として定義される。 したがって、プラスの極性(H2よりもイオン化傾向が小さい、あるいは還元力がH2よりも弱い)もマイナスの極性(イオン化傾向大、還元力強)も有する。 しかし、この電極電位から誘導される電池の起電力は、より電位の大きい方(これを貴という)から、より小さい方(卑と言う)を引いて得られる電極電位差であるから、常に正でなければならない。もし負であるとすると、それは電池ではない。
例を示そう。前のダニエル電池を反転して(Cu|Cu2+¦Zn2+|Zn)と表現し(3. 9)式から(3.8)式を引いたとすると、その電位差は-1.1Vで、
Cu + Zn2+=Cu 2+ + Zn (3.11)
となる。この反応は左から右へ自発的に進行する反応ではない(G>0)という 意味で、電池反応とは呼び得ない。(3.1)式(-ΔG=nFEcell)における、左辺のマイナス符号の意味に納得されよう。
以上述べてきた電極電位および電位差の極性に関する議論は、半反応を電子が反応系にあるように(Ox+ne=Red)書くことを前提としている。最近の酸化還元電位の表では、言うまでもなくそうなっている。 もし、Red=Ox+neのような逆転させて表現すると、極性もまた反転し、これまで述べてきた極性に関する約束事が目茶苦茶になってしまう。極性に関する混乱を招かないためにも、逆転させた表現はとらないことが望ましい。
3.1.4 電池活物質の酸化還元化学
実用電池には、 ⅰ)小型・軽量(単位体積或は単位重量当たりのエネルギー密度が高い)で、強力(出力密度(power density)が大きい)であることと、 ⅱ)保存性或いは電力貯蔵性に優れること、 ⅲ)エネルギー変換効率の高いこと、 ⅳ)安全性、信頼性、経済性、に富むことなどが要求される。自発的に進行する酸化還元反応は、元物質の組み合わせは極めて限られている。 それでは、どのような物質が実用電池(practical battery)に用いられているであろうか。この問題を酸化還元化学の観点から捉えてみよう。
<電池電圧と活物質の安定性>
正極反応に関与する反応物質(酸化剤あるいは酸化体、Ox)を正極活物質またはカソード活物質という。負極反応に関与する反応物質は負極(アノード)活物質である。 図3.3に水溶液系での実用電池(実用電池として開発中のものも含む)における活物質の組み合わせを標準電極電位の値と共に示してある。 まず、いわゆる使い捨て型の一次電池(primary battery)に注目してみよう。ダニエル電池は実用電池とはみなされないが、前節でも触れたので参考のため示してある。 正極活物質が貴に、負極活物質が卑にあり両者の電位差がEcellである。
今日、実用電池と呼ばれるもののほとんどが、正極活物質には金属酸化物を、負極活物質に亜鉛を用いていること、電解液にはアルカリ溶液(KOH)を用いること、 電池の名称に正極活物質の金属名を利用していることなどがわかる。例えば、酸化水銀―亜鉛電池(Ecell=1.35V)を水銀電池、 二酸化マンガン―亜鉛電池をマンガン乾電池(塩化亜鉛電解液使用)およびアルカリマンガン電池(アルカリ電解液使用)、 酸化銀―亜鉛電池(Ecell=1.59V)を酸化銀電池と呼称している。空気中の酸素を正極活物質に用いる酸素―亜鉛電池(Ecell=1.65V)は、空気電池と呼ばれる。
はじめに、なぜこれらの酸化物でなければならないかを検討してみよう。空気電池の正極反応の位置に注目していただきたい。この半反応の標準電極電位は、
E゜(O₂+2H₂O+4e=4OH-)=0.401V
である。逆反応はOH-イオンの酸化であるから、この電位よりも上位にある(より貴にある)正極活物質はOH-(すなわちH₂O)を酸化して、 それ自身はより低級の金属酸化物に還元される可能性がある。活物質が溶媒と反応してしまうのも自己放電の一種であり、避けなければならない。 逆にE゜(O₂/OH-)よりも下位(卑)にある活物質は安定であるから、E゜(O₂/OH-)よりも卑で電位的により近いところにある活物質ほどEcellがより大きくなり、 正極活物質として有利である。したがって、次の電位をもつAg₂OとMnO₂
E゜(Ag₂O+H₂O+2e=2Ag+2OH-)=0.345V,
および、 E゜(MnO₂+H₂O+e=MnOOH+OH-)=0.188V
の二つが、信頼できる正極物質として多用される理由が理解できる。
HgO(E゜(HgO+H₂O+2e=Hg+2OH-)=0.098V)は前の二つよりも安定な活物質であるが、Ecellは低くなる。
負極活物質についても、正極活物質と同様に溶媒との酸化還元化学からその安定性を評価できる。アルカリ中でのH₂Oの還元電位は、E゜(2H₂O+2e=H₂+2OH-)=-0.828Vである。 したがって、これよりも卑にある負極活物質はH₂を発生して自己溶解する可能性を有し、安定な活物質とはなり得ないはずである。これは熱力学的な一般則である。 ところが活物質の亜鉛は、E゜(Zn(OH)₄2-+2e=Zn+4OH-)=-1.211Vもの卑な電位を有するにも関わらず、実用電池の負極活物質として欠かせない電池材料となっている。 これはなぜであろうか。高純度の亜鉛は水素発生の起こり難い金属として知られている。後でも述べるように、 このような現象を水素発生の過電圧(overpotential)が非常に大きいと表現する。すなわち、熱力学的には水素発生が起こるはずであるが、 速度論的にその発生速度が極めて小さいという意味である。実際の電池では、水素過電圧(hydrogen overpotential)をさらに大きくするために、 亜鉛よりもさらに水素過電圧の大きい水銀を微量亜鉛表面に塗布(アマルガム化)してある。こうすることにより、使用期間内での亜鉛の自己放電は実質は例外的な負極活物質であり、 今日の実用一次電池が1.5Vもの開路電圧(熱力学的には水の分解電圧である、1.23V止まりである)を持ち得るのは、亜鉛の水素過電圧のお陰であると言っても過言ではない。
これまで述べてきた実用電池の活物質はすべて個体であって、溶媒に難溶性の化合物または単体が用いられている。 アルカリ電解液を用いるのも酸化物活物質を溶解させないためである。さきにセパレーターが電池構成上の要素材料として欠かせないものであることを述べたが、 活物質がダニエル電池のそれのように溶解型であるとすると、セパレーターを通じての活物質の混合が起きるから、安定な電池を構成できない。 両極活物質が固定であるというのも、製品としての長期安定性を要求される一次電池の特徴の一つである。
次に、二次電池(secondary cell)の活物質の化学についても簡単に触れておこう。二次電池は繰り返し使用(放電⇄充電)の可能な電池であり、 十分な充放電サイクル数の確保と、水溶液系では過充電時に発生する水素および酸素に対する対策が要求される。まず、最もポピュラーな鉛電池についてみてみよう。 鉛電池の特徴は電解液が酸(硫酸)であることと、開路電圧が2.04Vと異常に大きい活物質の組合わせである。特に後者は、水の分解電圧が1.23Vであることを考えると、 実に信じられない高電圧である。このような高電圧を維持できる秘密はなんであろうか。負極活物質であるPbの電位は、E゜(PbSO₄+2e=Pb+SO₄2-)=-0.395Vであり、 Znの場合と同様にH₂を発生して自己溶解してもおかしくない位置にある。しかし亜鉛と同様にZn、Hg、Pbは電池負極材料の御三家ともいうべきで、 ちなみにそれらの水素発生の交換電流密度は、他の金属材料、例えばPtに比べて106~108分の1にすぎない(第2章の2.3.5 ⅱ)表2.10参照)。
PbO₂の正極電位は、E゜(PbO₂+SO₄2++4e=PbSO₄+2H₂O)=1.682Vであり、酸性中でH₂Oの酸化(E゜(O₂+4H++4e=2H₂O)=1.23V)を引き起こすに充分な強い酸化剤である。 一般にO₂発生の過電圧はH₂発生のそれに比べてかなり大きく、アルカリ中でE゜(O₂/OH-)よりもやや貴にあるNiOOHやAgOが活物質として使用できるのは、このためである。 しかし、酸性中のPbO₂はO₂発生の過電圧が特に大きいものの一つとして知られており、このような極めて貴な電位にあってもPbO₂自体の安定性は確保されている。 このように鉛電池はPbO₂のO₂発生過電圧の大きさの双方の効果が加わって、水溶液系の電池としては異例ともいうほど高い電圧を保持できる。このような活物質の組合せは、 今のところ他にない。鉛電池は100年以上の歴史を持つが、今なお二次電池の主役の座を占めている理由がここにある。
ニッケル・カドミウム電池もユニークな活物質の組み合わせをとっている。Ecellを大きくするという観点からすれば、負極活物質として亜鉛を用いる方が良さそうにも思える。 しかし、亜鉛負極の二次電池化は極めて困難とされている。その理由として、充電時における亜鉛の析出形態が樹枝状(デンドライト)形態をとり、内部短絡や亜鉛結晶の剥離を生じて、 充放電サイクル数を大きくできないことが挙げられている。Zn(Ⅱ)は濃厚アルカリ中でオキシアニオン(Zn(OH)₄2-, ZnO₂2-)として溶出するところに問題がある。
カドミウムの電位は、E゜(Cd(OH)₂+2e=Cd+2OH-)=-0.809Vで、アルカリ中でのH₂Oの還元電位より貴にあるからCd自体は安定である。 正極活物質NiOOH(E゜(NiOOH+H₂O+e=Ni(OH)₂+OH-)=0.52V)との電池反応は次のようである。
2NiOOH+Cd+2H₂O=2Ni(OH)₂+Cd(OH)₂ (3.12)
Ecellは約1.3Vで鉛電池のそれよりははるかに低いが、鉛電池よりも長寿命で信頼性が高い。それは ⅰ)鉛電池のように電解質イオン(SO₄2-)が反応に関与しないこと、 ⅱ)電池式から明らかなように充放電に際して水の生成(充電時)と消滅(放電時)しか起こらないこと、 ⅲ)後に述べるように過充電時のガス発生(H₂とO₂)を抑える巧妙な方策がとられていて、メインテナンスフリーまたは完全密封化が可能なことなどによる。
金属材料の安定性を調べることは、その環境で金属、酸化物、水酸化物、イオン等のいずれが熱力学的に最も安定かを検討することに対応する。通常の環境では腐食に関与する最も重要な因子は水であることから、水溶液中における金属の熱力学的安定性が広く調べられている。
まず、亜鉛を例にその安定性を考えてみよう。亜鉛の水酸化物(酸化物)、イオンとしてそれぞれ Zn(OH)2、Zn2+を考えることにする。Zn/Zn2+については、Zn2+の標準化学ポテンシャルロがμ°、-147.1kJ/molであることから
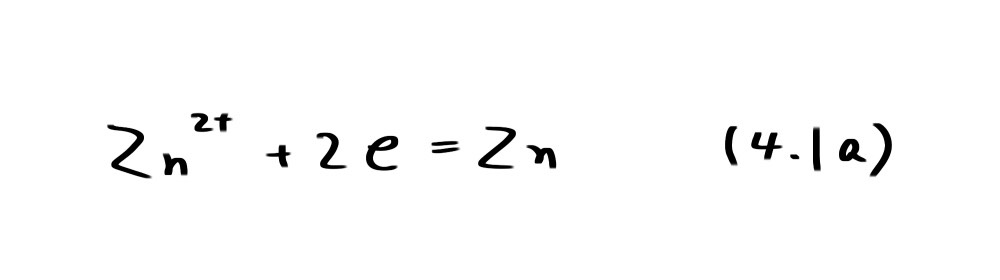
の反応の平衡電位は Nernst 式から次式となる。
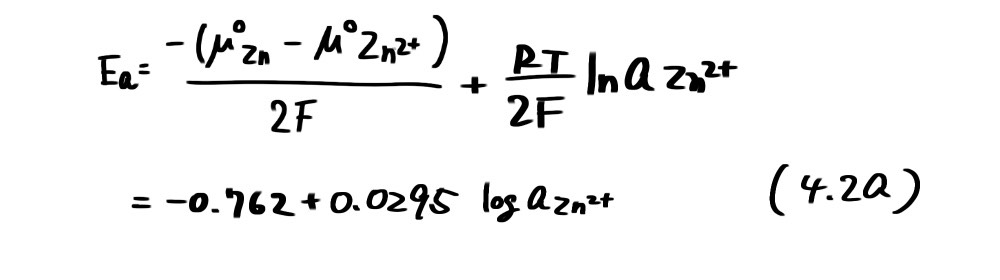
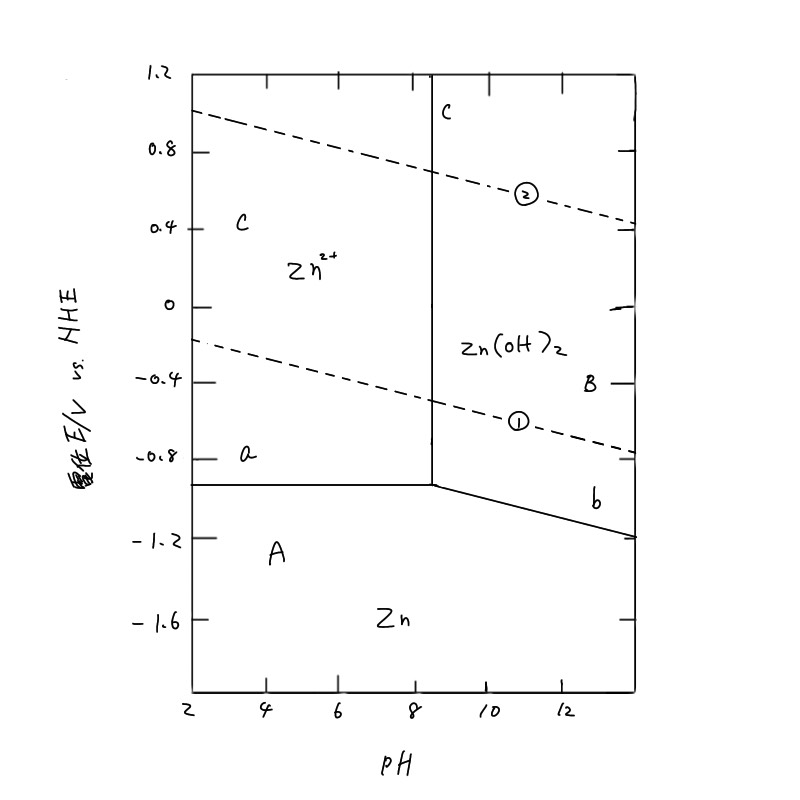
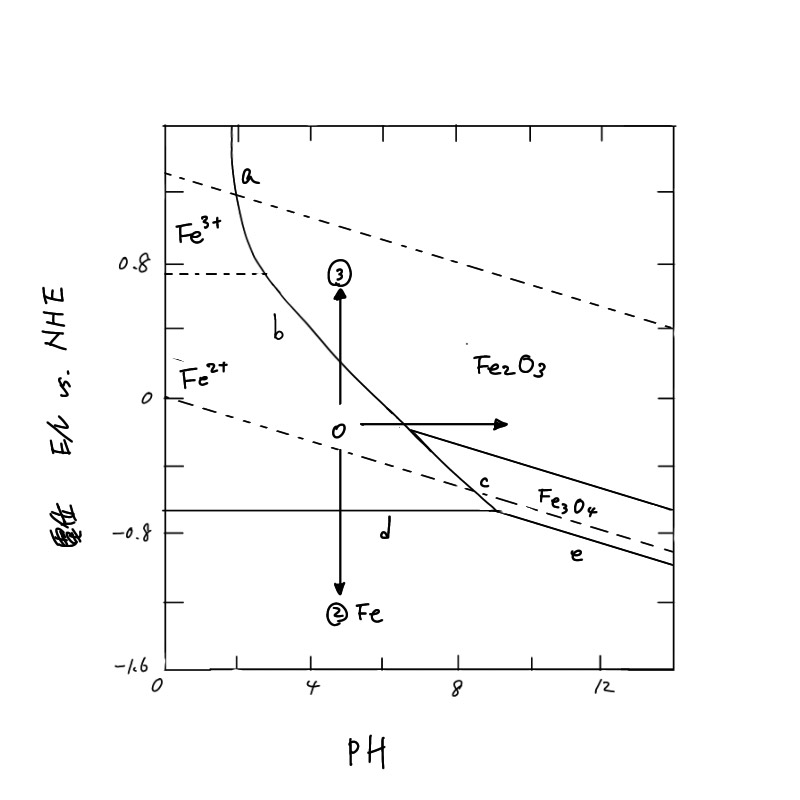
例えば、Zn2+の活量aZn2+がその濃度と等しく10-6mol/lであったとき、Ea=-0.939Vとなり、電位がこれよりも高くなるとZn2+の濃度が増加する。すなわちZnの溶解が起こる。(4.2a)は電位-pH図(potential-pH diagram)(図4.1)で直線a で表わされ、この直線より上ではZn2+が、下ではZnが安定であることを示している。
Zn/Zn(OH)2は、Zn(OH) 2とH2Oのμ°がそれぞれ-553.6および-237.2kJ/molであり、Zn(OH) 2は固相として存在するためその活量aZn(OH) 2=1と考えれば
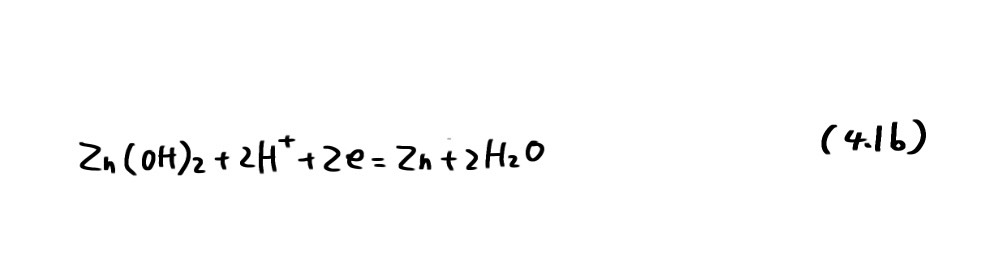
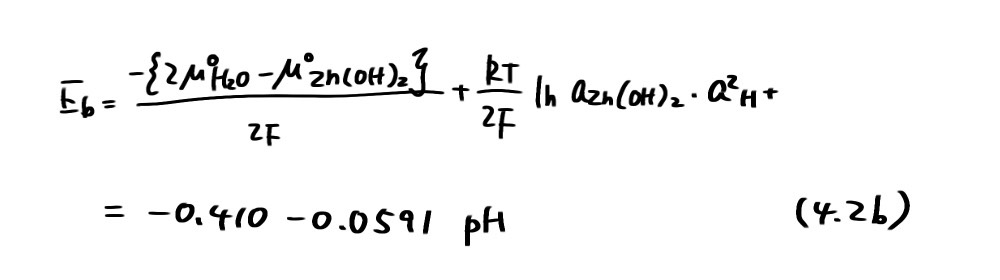
となり、平衡電位はpHに依存する。(4.1b)の反応は平衡電位Ebより高い電位では左向きに、低い電位では右向きに進行する。すなわち、図4.1の直線b より高い電位ではZn(OH) 2が、低い電位では Zn が安定な領域である。 Zn2+ /Zn(OH) 2の平衡反応は
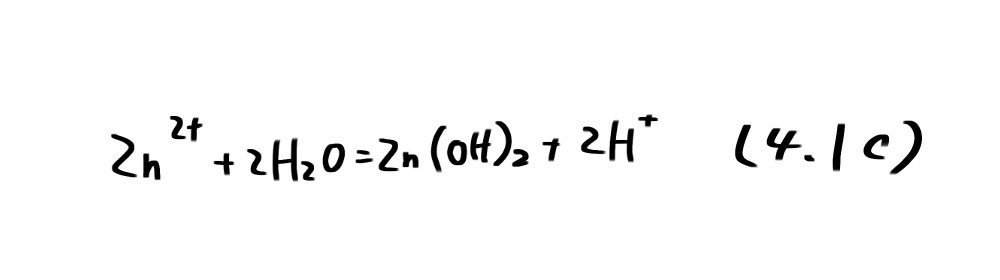
の沈殿平衡反応であり、電子の授受が無いため電位に依存しない。Zn(OH) 2の溶解度積Ks=aZn2+・a2H+=1.80 × 10-14、Kw= aH+・aOH-/aH2O=10-14を用いれば、この反応の平衡定数Kcは
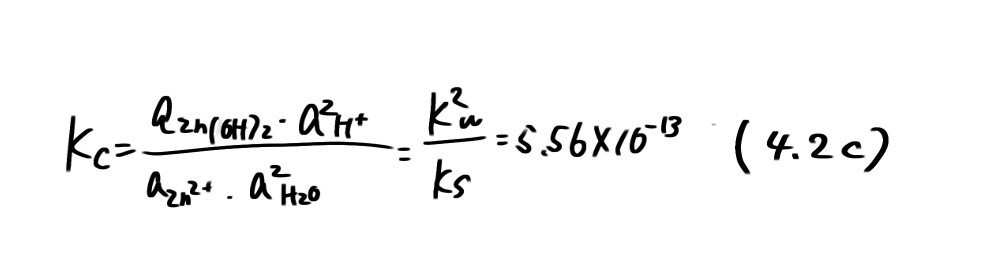
で表わされる。Zn(OH)2とH2Oの活量を1とおけば
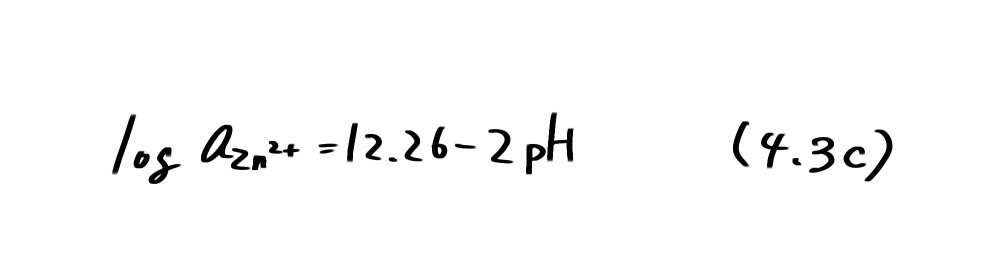
となり、pHの変化で溶解するZn²+の濃度が増減することを示している。例えば azn2+ = 10-6を基準として、pH=9.13 (図4.1の直線c)より低pH では Zn(OH) 2の溶解度が増加して、(4.1c)の反応が右に進み、これより高 pHでは左に進むことを示している。いいかえれば、Zn2+では低 pH 側で、Zn(OH)2は高 pH 側で安定であることを示している。以上のことから、図4.1の実線で区切られた3つの領域A、B、Cはそれぞれ、Zn、Zn(OH) 2、Zn2+安定領域であるといえる。Aの領域は金属Znで腐食が起こらないことから不活性態 (immunity)あるいは不感域と呼ばれ、Cの領域は Zn2+として溶解が起こることから腐食域と呼ばれる。一方、Bの領域はZn(OH)2が安定で、Znの酸化は進行するが、金属表面を酸化物あるいは水酸化物が覆いつくしてしまうと、その後の酸化物の成長は極端に遅くなるため、実質的に腐食反応が停止したのと同じになる。このような状態を不働態(passivity)と呼び、腐食が進行しない領域と考えることができる。図4.1では低pH側だけについて示したが、アルカリ性ではZn(OH)2がZnO22-として溶解し、その溶解度が増加するため再び腐食域が現われる。
図4.1には亜鉛の安定性ばかりでなく、水の安定領域も示してある。図中の破線①は次に示す水素発生の反応の平衡電位(水素分圧 PH2=l atm)である。
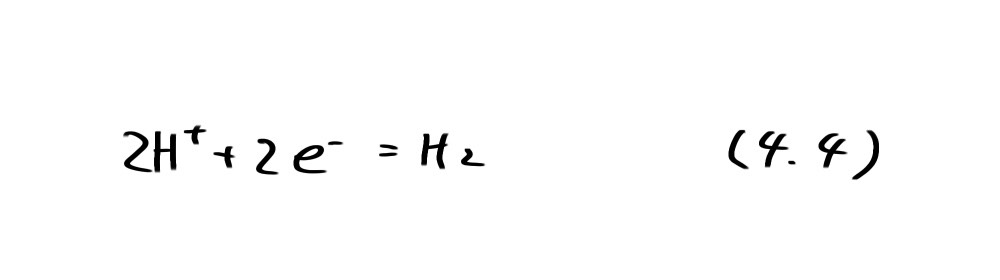
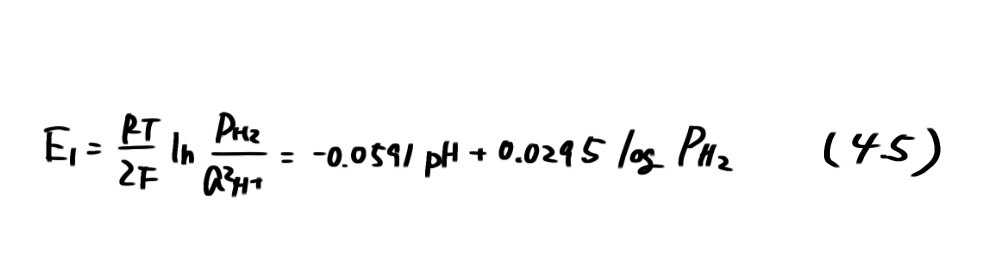
すなわち、直線①以下では水素分圧が1atm 以上になり水素発生が起こることを示している。一方、破線②は次の反応の平衡電位(酸素分圧PO2=l atm)であり
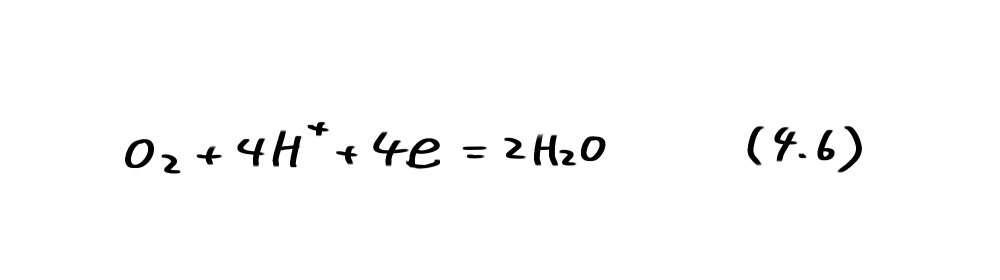
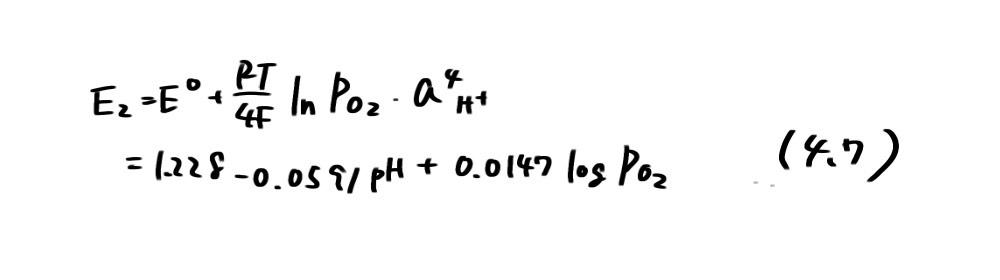
で表わされる。直線②より上ではPO2>l atmとなり、水が分解して酸素ガスが発生すること、直線②より下では溶液中に溶け込んでいる酸素が還元されることを示している。
さて、金属材料の腐食では、金属が自発的に(外部から電流・電圧を与えることなしに)溶解し、または酸化物を形成する反応が進む。図4.1では金属の酸化反応(アノード反応)に注目して、腐食域などの領域を決めた。しかしながら、このアノード反応によって生じた電子を消費するカソード反応が同時に、自発的に起こらなければ全体的な腐食反応は停止してしまう。通常の環境で考えられるカソード反応は水の関与する反応である。 たとえば、酸性溶液中での亜鉛の腐食は
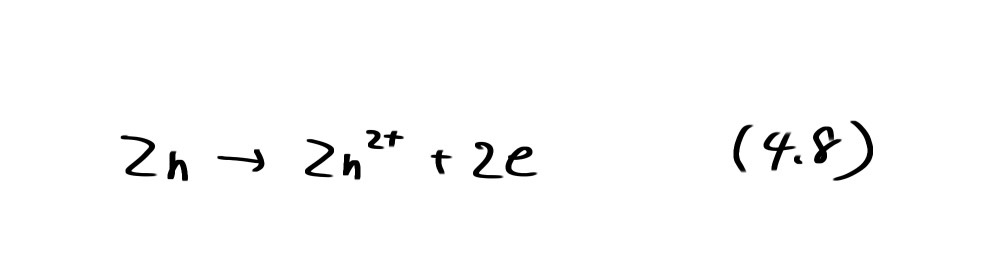
のアノード反応と水素発生または酸素還元のいずれかのカソード反応が起こる。
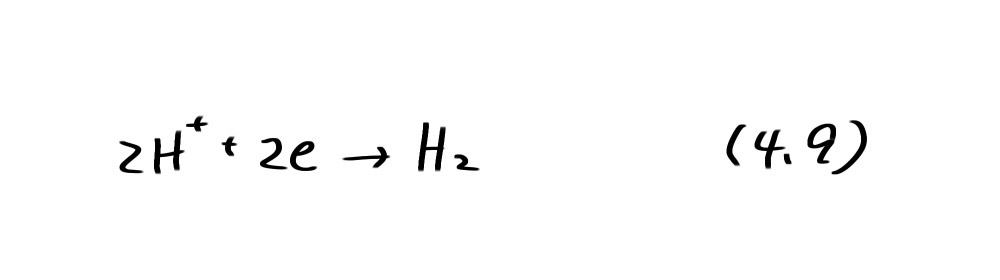
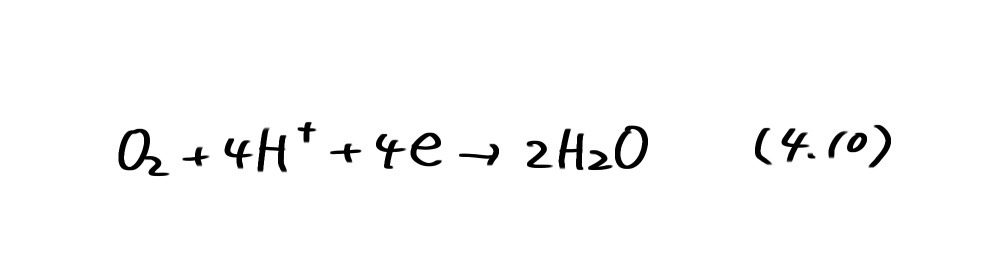
アノード、カソード反応共に自発的に起こることが必要である。それ故、亜鉛が水素を発生しながら腐食するのは図4.1のA領域の破線①より下の部分(水素発生型腐食)、容液に溶け込んだ酸素が還元されて亜鉛が溶解するのはA領域の破線②より下の部分(酸素消費型腐食)である。
腐食するか、あるいは不感態、不働態として安定に存在し得るかは、電位-pH図によっておおまかな判定が可能である。例えば、pH5.0の環境におかれた鉄の電位が-0.20V であったとする。この状態は鉄の電位-pH図 (図4.2) において腐食域にあるが、水素発生型の腐食は起こらず、溶液中の酸素が還元される酸素消費型の腐食が起こることがわかる。また、腐食を防止するためには、酸化剤である酸素を除去することのほか、図中の矢印で示された3つの方法が考えられる。➀は環境のpHを高くし不働態にする方法、②は外部から電圧をかけカソード分極して不感態にする方法(カソード防食法, cathodic protection)、そして③は同様にアノード分極して不働態にする方法(アノード防食法, anodic protection)である。
電位-pH図は水溶液環境における金属の熱力学的安定性をわかりやすく表現しており、Pourbaixによって大部分の金属元素について集大成されたことからプールペイ図(Pourbaix diagram)ともよばれている。しかしながら、この図を使用する場合には次のことに留意しておく必要がある。
➀速度に関する情報;電位-pH 図は熱力学的安定性だけを議論したもので、速度に関する情報は含まれていない。
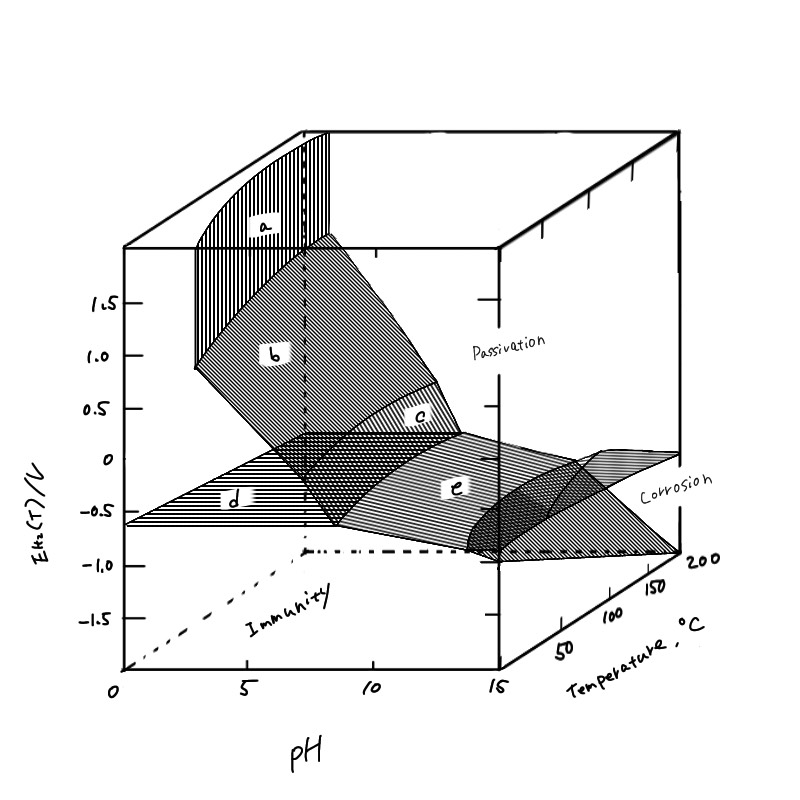
図4.3 鉄一水系の電位-pH-温度図
図中のa~eの面はそれぞれ図4.2のa~eの直線に対応する。
(H. E. Townsend, Jr.3 Corros. Sci., 10,434(1970)より)
②温度、電解質の影響;亜鉛の例で計算に用いた平衡反応の熱力学定数は温度の関数であることから、温度が異なれば電位-pH図も変化する。図4.3は鉄の電位-pH―温度の関係を三次元で表わしたもので、温度上昇とともにアルカリ側の腐食域が若干拡大している。また、電解質中に金属イオンと錯体を形成するイオン(例えば、CN-、NH4-、CT-など)が含まれる場合には、酸化物、金属の安定領域がせばまり腐食域が拡大する。特に塩化物イオンが含まれる場合には酸化物が局所的に不安定になり、孔食などの局部腐食が発生するが、電位-pH 図の熱力学的安定性だけからその発生を予測することは困難である。
③合金元素の影響;合金中の各元素の活量、複合酸化物の形成など複雑な問題があるため、それぞれの合金成分元素の安定性を重ね合わせて、合金全体の安定性を論ずる場合が多い。
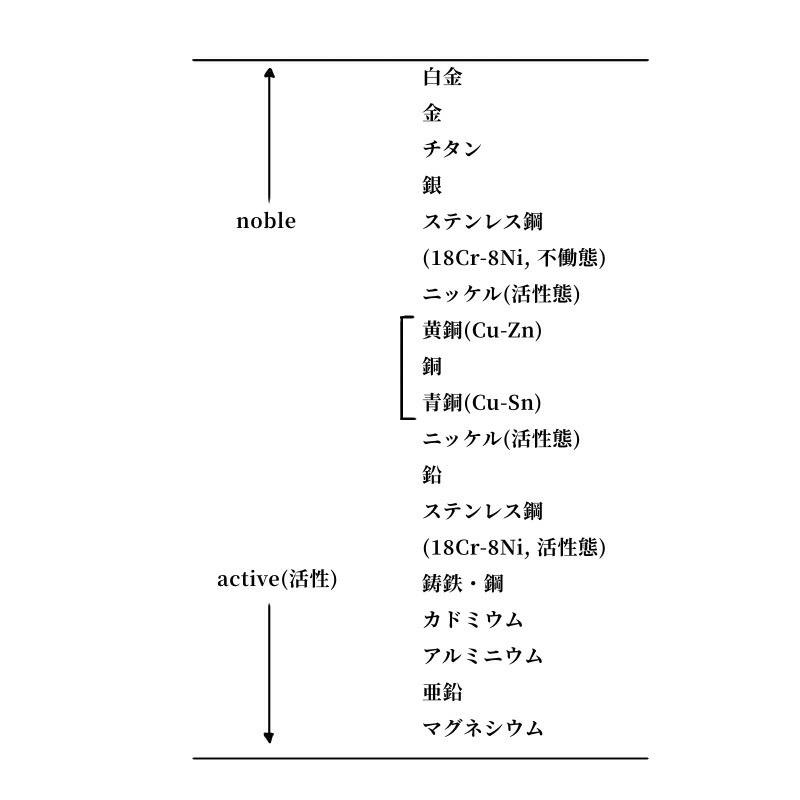
純粋な金属とその金属イオンの水溶液における標準電極電位は、その金属に特有な値であり、標準生成自由エネルギーから計算される。表4.1はいくつかの金属について、電位が貴(noble)な順に並べたもので、金属のイオン化系列と逆順になっている。この表で上にあるほど水溶液中で安定であり、下にいくほど不安定であるといえる。また、電池の章で述べられているように、この表から2組の可逆系を選び電気化学的に接続すると、電池が構成され、卑(less noble) な金属の溶解と貴な金属の析出が起こる。
表4.1 水溶液系におけるいくつかの金属の標準電極電位
(標準水素電極 (NHE) 電位基準、25℃)

実用的な金属・合金の海水中における電極電位の系列を表4.2に示す。この表では、不働態状態と活性状態の電位が示されており、表の下にある材料ほど腐食しやすい。異種の金属・合金を接続した場合も上に述べたと同様な腐食が起こるが、これについては後の節で腐食速度を含めて詳しく検討する。
金属材料を腐食から守るためのもっとも単純で分かりやすい方法は、材料を腐食性環境から遮断することである。その意味で古くから金属の表面を種々の材料で、例えば結城塗料、無機物のほうろうあるいは金属によるめっきなどにより被覆することが行われてきた。また,「腐食の平衡論」(4.1)で述べたように環境との平衡条件を変えること、環境または材料の腐食反応の特性を変えることによる防食も考えることができる。ここでは、塗装・被覆以外の方法によるいくつかの防食法について考えることにする
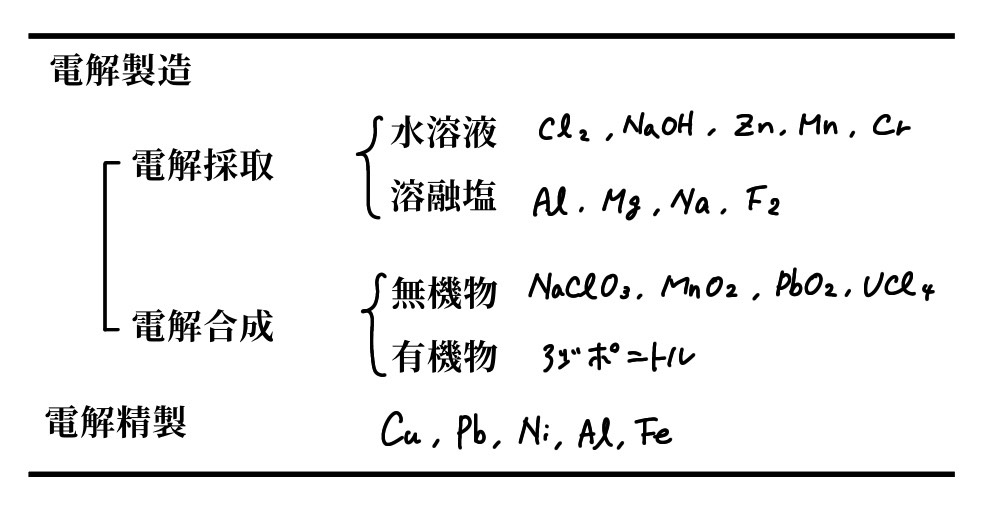
工業電解(industrial electrolysis)プロセスとは電気化学システムを用い、電子伝導体である電極とイオン伝導体である電解質との界面で起こる電極反応を利用して、より質の高い物質の製造、材料の表面処理を行うプロセスのことである。ここでは投入される電気エネルギーが物質の化学エネルギーに変化する。広義ではメッキ、電解加工等の表面処理、さらに二次電池の充電過程も含まれるがこれらは別項に譲るとして、この章では狭義の工業電解プロセス、すなわち物質を作り出すための電気分解のプロセスについて取り扱うことにする。
工業電解プロセスは目的によって大きく2種類に分類できる。一つは物質の製造に直接電解反応が利用されるもので食塩電解の塩素、アルミニウム製錬のアルミニウムがその代表的なもので電解製造と呼ぶことができる。ここでは金属の電解採取(electrowinning)の他に、無機、有機、の製品の合成に用いられることもあり、これらは電解合成(electrosynthesis)と呼ばれるとこともある。他の一つは物質の純度を高めるために電気分解を利用するもので銅を始めとする金属の電解精製(electrolytic refining)がその代表的なものである。表5.1にはその分類と代表的な製品を示す。
電気分解を行う装置を電解槽(electrolytic cell)あるいは高温溶融塩では電解炉という。基本的にはアノード(電解プロセスでは陽極)、カソード(陰極)の二種類の電極、電解質、隔膜の4つの要素から成り立っている。このうちも隔膜は無い場合もあるが二 種の電極、電解質は必ず必要であるアノードでは脱電子反応すなわち酸化反応が、カソードでは脱電子反応すなわち還元反応が起こる。電解質中には反応に関与する物質がイオンまたは分子の形で存在しており、原料の供給、製品の輸送の役割も果たす。隔膜はアノード生成物、カソード生成物の分離のために必要に応じて利用されている。表5.2にはこれらの様式を分類して示す。

電解質としては酸またはアルカリといった水溶液が最も多く利用されている、また水溶液あるいは常温で固体の塩を高温で融解させた溶融塩を用いるのは水溶液中では不可能な、例えば酸素発生より貴な電位の反応、あるいは水素発生より卑な電位の反応を行わせる際に用いられる。固体電解質とは固体の状態で、イオン伝導を有する物質 をいい、1,000℃付近の高温でのO2-イオン導電体のジルコニア(ZrO2)、H+イオン導電体の高分子固体電解質(SPE)などがある。これらは電解質であるとともに隔膜の機能も有しており、これからの電解プロセスへの応用が期待されている。ろ隔膜では基本的にアノード液とカソード液はつながっているが、この膜を用いることにより両者の混合をある程度防ぐことができる。密隔膜ではアノード液とカソード液は完全に分離されており、膜中はイオ ンが選択的に通過する。
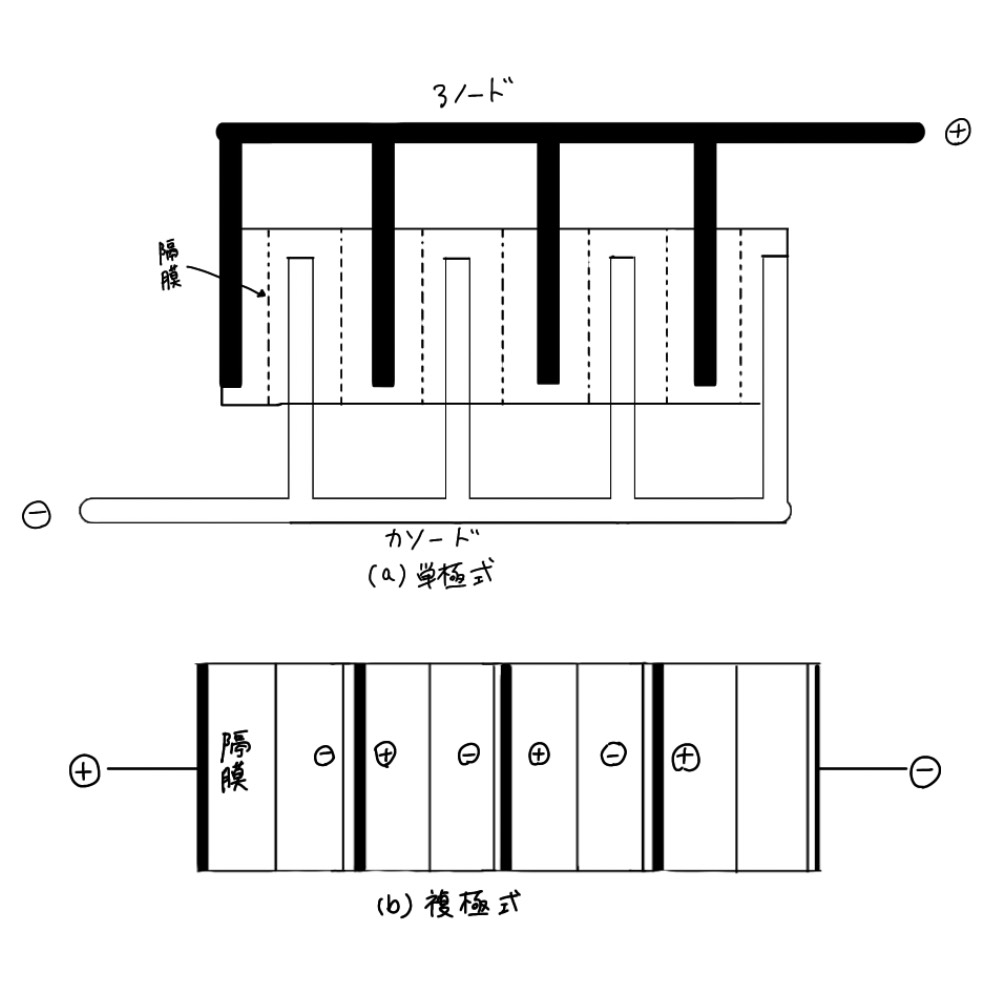
電解槽中で反応は電極と電解質の界面で起こるので、反応速度(生産量)は電極面積に比例する。多孔性電極を用いて電極面積自体を大きくする方法もあるが、通常は電解槽を積み重ねて工場の単位面積当りの反応面積を拡大する方 法が用いられている。 水溶液を用いる電解においてはこの電解槽の電極接続方法に単極式と復極式の二種があり、それぞれ単極式電極(monopolar electrode)、複極式電極(bipolar electrode)と呼ばれるが、図5.1にこの接続様式を示す。単極式は低電圧、大電流の様式であり、電解槽電圧は単槽電圧に等しいが電極 に電気を供給する導体(ブスバー)が多量に必要となり、ことでの電圧のオーム損が大きい。複極式では一つの電極が片側がアノード、反対側がカソード と二つの役を担っており、槽電圧は単槽電圧 X槽数となるので単極式に比べて大電圧、小電流の様式である。ここでは槽間の接続ブスバーは不必要でそのオーム損は小さくなるが液を通しての短絡電流を防ぐなどの工夫が必要となる。
工業電解プロセスは幅広く応用されている。世界中で生産されている単一元素の純粋な金属あるいは非金属単体の製造方法について図5.2で示す。周期率表の各元表に重ねて環が描かれている。電解採取により製造されている単体の割合を、環の黒く塗られた部分で示す。調査可能であった84元 素のうち、60%以上の52元素が直接電解プロセスを経て生産されている。そのうちナトリウム、フッ素、塩素のように電解採取以外に製造法のない単体や、アルミニウム、銀、白金のように経済的な
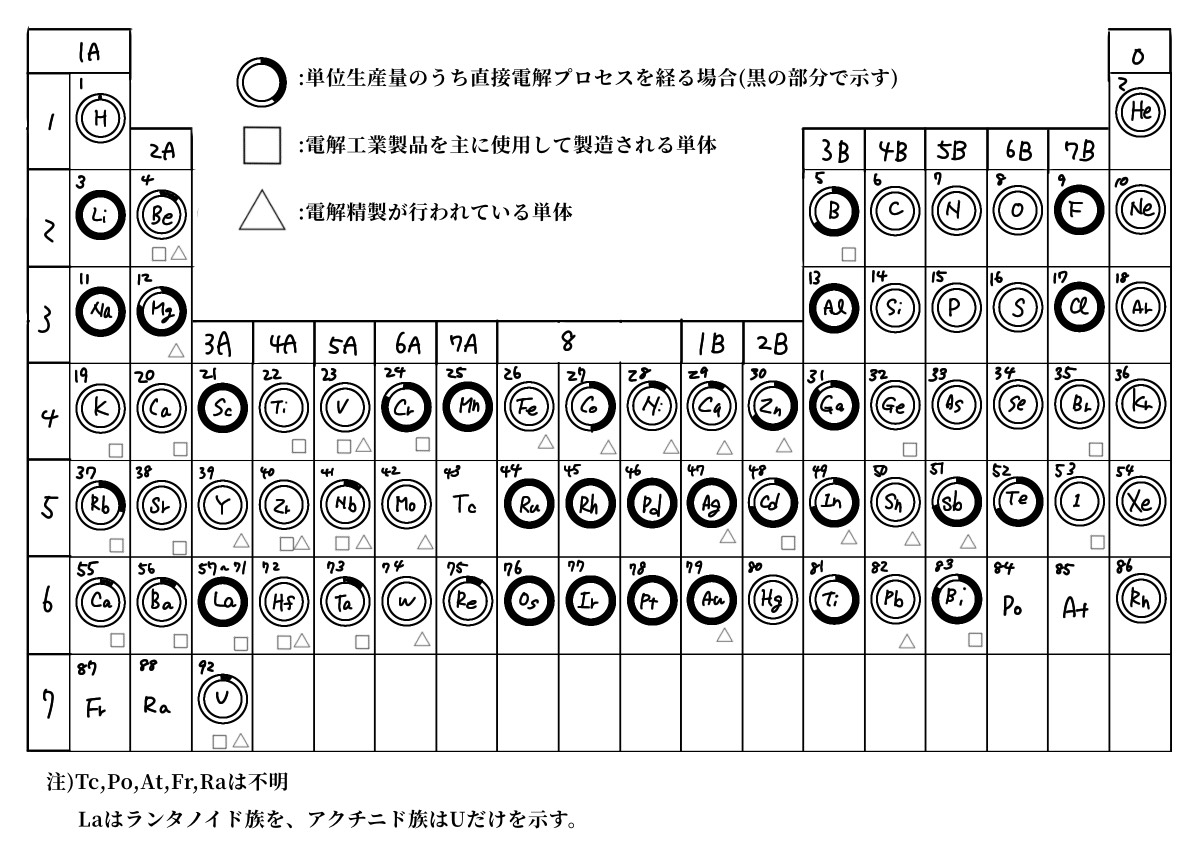
有利さから、全量電解採取によって製造 されている単体を合わせると16元素にものぼる。カリウムやチタンのように直接の電解採取ではないが、電解採取によって製造されるナトリウムやマグネシウムの強い還元力を使って生産しているものもある。このような単体の元素には□印をした。結局アルカリ金属、アルカリ土類金属、ハロゲンはすべて直接・間接に電気化学的な方法に依存している。また鋼のように精製過程で電気化学的方法が重要な場合もある。これには△印をつけた。この図で無印の元素、つまり電気化学的方法とはまったく関係のない元素は、たった15元素である。80%以上の種類の単体の製造に工業電解プロセスが使われていることになる。これらの工業電解プロセスの特徴は次の通りである。
i)熱エネルギーだけでは進まない、反応を行わせることができる。
ii)ファラデーの法則に従い、生産量は流した電気量に比例する。
iii)大電流容量で低電圧の直流電気エネルギーを利用する。
iv)電圧を調整することによる副反応の発生を制御できる。また電流を調整することにより生産量の調整ができる。
v)酸化反応、還元反応が別々の場所で行われるため二種以上の製品が得られ、各々高純度のものが得られる。
vi)反応が電極界面に限られる欠点がある。このため一つの反応容器による生産速度が限られ、大量生産のためには同じ容器を多数必要とする。
電気分解に必要な電気エネルギーは(電圧)×(電気量)の積の形で表わされる。 理論的な最小電圧は物質の持つ化学エネルギー値から計算される。理論分解電圧に等 しい。電気量に関してはファラデーの法則により理論的に定まる。いま総ての電流 が製品Jの製造にだけ使われたとして、生成した製品の重量 wj。、jの式量Mj、nj。 をjのモル数、νjを化学量論係数、nを反応に関与する電子数、Fをファラデー定 数、qを通過した電気量とすると次式が成り立つ。
d wj。=Mjtnj。=(νjMj/nF)dq (5.1)
Q0=nF/νjMj (5.2)
ここではQ0は反応種と反応式が決まれば定数となり、理論電気量と呼ばれる。実 用単位としてkAht-1または Ahkg-1で表わされることが多い。電解に必要な電気量 もこれを下回ることはできない。表5.3には無機工業電解で生産されている物質の いくつかとその理論電気量を示す。 実際の操業では理論値で稼働することはない。まず電気量について示すと、(実 際に得られた目的の製品の量wj)/(流れた電気量から計算される製品の理論生産量 wj。)を電流効率εFと呼んでおり、単位重量を得るのに実際に流れた電気量Qと理論電気量Q0との比になる。| 物質名 | 式量(g・mol-1) | 理論電気量Q0(kA・h・t-1) | 反応電子数z/y | 種別*2) | 成分(条件)*2) | 主反応物 |
|---|---|---|---|---|---|---|
| 銀 | 107.88 | 248 | 1 | aq | AgNO3+Cu(NO3)2 | 粗Ag → 精製Ag |
| アルミニウム | 26.96 | 22980 | 3 | aq | AlF3・3NaF(1,000C) | 2Al20+3C=4Al+3CO2 |
| 金 | 197.0 | 408 | 3 | aq | HAuCl4+HCI | 粗Au→ 精製Au |
| ビスマス | 209.00 | 385 | 3 | aq | BiCl3+HCI | 粗Bi → 精製Bi |
| カルシウム | 40.08 | 1338 | 2 | f | CaCl3 (800C°) | CaCl2=Ca+Cl2 |
| カドミウム | 112.41 | 477 | 2 | aq | CdSO4+H2SO4 | 2CdS04+2H20=2Cd+2H2SO4+O2 |
| 塩素 | 70.91 | 755 | 2 | aq | NaCI(D) | 2NaCI+H2O=2NaOH+Cl2 + H2 |
| クロム | 52.01 | 1546 | 3 | aq | Cr2(SO4)3+(NH4)2SO4(D) | 2Cr2(SO4)3 + 6H2O=2Cr+6H2SO4+2CrO2, |
| クロム酸 | 100.01 | 803 | 3 | aq | Cr2(SO4)3 | Cr2(SO4)3+6H20=2CrO3+3H2 +3H2SO |
| 銅 | 63.54 | 844 | 3 | aq | CuSO4+H2SO4 | 粗Cu→ 精製Cu |
| 銅 | 63.54 | 422 | 2 | aq | CuCl+NaCl | 粗Cu→ 精製Cu |
| フッ素 | 38.00 | 1410 | 2 | f | KF・HF (1000C°) | 2HF=2H2+2F2 |
| 鉄 | 55.85 | 960 | 2 | aq | (NH4)2SO4> FeSO4 | |
| ガリウム | 69.72 | 1153 | 3 | aq | Ga(OH)3+NaOH | 4Ga(OH)3=4Ga+302+6H20 |
| 水素 | 2.016 | 26587 | 2 | aq | KOH | 2H2O=2H2+O2 |
| インジウム | 114.82 | 700 | 3 | aq | InCl3 +NaCl | 2InCl3=2In+ 3Cl23 |
| リチウム | 6.94 | 3862 | 1 | f | LiCI+KCI (450C°) | 2LiCl2=2Li+ Cl2 |
| マグネシウム | 24.32 | 2204 | 2 | f | MgCl2 +CaCl2+ NaCI (700C°) | MgCl2=Mg+Cl2 |
| マンガン | 54.94 | 976 | 2 | aq | MnS04+(NH4)2SO4 | 2MnSO4+2H2O=MnO4+2H2SO4+02 |
| 二酸化マンガン | 86.94 | 617 | 2 | aq | MnSO4 | MnSO4+2H2O=MnO2+H2SO4+H2 |
| ナトリウム | 22.99 | 1166 | 1 | f | NaCl+CaCl2 (560C°) | 2NaCI=2Na+Cl2 |
| 塩素酸ソーダ | 106.45 | 1510 | 6 | aq | NaCl | NaCl+ 3H2O=NaClO3+3H2 |
| 過塩素酸ソーダ | 12.45 | 438 | 2 | aq | NaClO3 | NaClO3+H2O=Naciou +H2+ Cl2 |
| 水酸化ナトリウム | 40.0 | 670 | 1 | aq | NaCl+NaOH(D) | NaCl+H2O=2NaOH+H2+Cl2 |
| 過硫酸アンモニウム | 228.21 | 235 | 2 | aq | NH4HSO4 | 2NH4HSO4=(NH44)HS2O8+H2 |
| ニッケル | 58.71 | 913 | 2 | aq | NiSO4+H3BO4+NaCl | Ni2S3+3S |
| 鉛 | 259 | 2 | aq | PbSiF4+H3SiF4 | 粗Pb → 精製Pb | |
| スズ | 118.70 | 903 | 4 | aq | NaOH+Na2SnO3 | 粗Sn → 精製Sn |
| 亜鉛 | 65.38 | 820 | 2 | aq | ZnSO4+H2SO4 | 2ZnSO4+2H2O=2Zn+2+H2SO4+O2 |
εF=Q0/Q (5.3)
この電流効率(current efficiency)の低下の原因は主に次の4つである。 i) 副反応の存在:食塩電解のアノードでの酸素発生反応がその例であり、電極触 媒の改良により改善できる。 ii) 不純物による電解電流の存在:銅の電解採取の際、電解液中に Fe3+/Fe2+ のレドックス対が不純物として存在し、アノードで酸化生成した Fe3+ がカソードで Fe2+にもどるサイクルを繰り返すので電流の損失となる。 iii) 電流の漏えい、短絡:電極以外の所で電流の出入りがあったり、電極の変形、 金属電解の際の樹枝状析出物による電極の短絡により電流効率は低下する。 iv) 生成物の分解あるいは再酸化:アルミニウム電解では生成したアルミニウムが CO2により再酸化されて電流効率が低下する。 次に電圧(cell voltage) について考えてみる。いま電流Iが流れている状態で 測定したアノード、カソードの電極電位をそれぞれEa、Ecとすると電解槽の全電圧 Vtは次式で表わされる。Vt=(Ea-Ec)+I(RL+RS+RD) (5.4)
ここで、RLは電極から電源までの導体抵抗、RSは電解質のオーム抵抗、 RDは隔膜に 起因する抵抗である。電解槽の反応面積の特性値を A*(これは必ずしも実際の面積 とは一致しない。)を用いて電流密度 J(=I/A*) で書き換えると次式になる。Vt=(Ea-Ec)+J(βL+βS+βD)
ここで、β=RA*である。さらに平衡電位(equilibrium potential) Eと過電圧 (overpotential)を用いると
Ea-Ec=(Ea。-Ec。)+(ηac) (5.6)
(Ea。-Ec。) は理論分解電圧 (theoretical decomposition voltage)Edと呼ばれ、これ 以下に電圧を切り下げることはできない。また目的の電解反応のギブスの自由エネ ルギー変化ΔG。 と次のような関係がある。W=Q×Vt=Q0Vt/εF (5.9)
また、理論電解エネルギー W0(=Ed×Q0)との比をエネルギー効率εw と呼ぶ。εw=W0/W=(Q0/Q)(Ed/Vt)=SVSF (5.10)
表5.4には代表的な工業電解プロセスのこれらの特性値の例を示す。電流効率と 比較して電圧効率が低い事が特徴的である。| プロセス | アルミニウム電解精錬 | 食塩電化イオン交換膜法 | 銅電解精製 | 亜鉛電解採取 |
|---|---|---|---|---|
| 理論電気量(kA・h/t) | 2980 | 670 | 844 | 820 |
| 理論分解電圧(V) | 1.17 | 2.2 | 0.1mV | 2.0 |
| 理論電解電力(kW・h/t) | 3490 | 1470 | 0.084 | 1640 |
| アノード電流密度(A/m(2) | 8000 | 3000 | 220 | 450 |
| 単槽電圧(V) | 4.0 | 3.15 | 0.31 | 3.3 |
| 電気量原単位(kA・h/t) | 3350 | 700 | 920 | 910 |
| 電解電力(kW・h/t) | 13400 | 2200 | 284 | 3000 |
| 電流効率(%) | 90 | 96 | 97 | 90 |
| 電圧効率(%) | 29 | 70 | 3.2×10-4 | 61 |
| エネルギー効率(%) | 26 | 67 | 3.2×10-4 | -55 |
水素は石油精製、アンモニア合成など広範な分野で用いられている。かつては水を電気分解して水素を得る水電解(water electrolysis)が重要な工業プロセスであったが、現在では、大部分石油、天然ガスなどの改質により作られている。特に電力コストの高い我国においては大規模なものはほとんどなくなった。しかい、世界的に見ると、カナダ、ブラジル、ザイールなどのように水力発電により安い電力の得られる箇所には大型新鋭電解槽が作られている。また、クリーンな二次エネルギーとして注目されている水素を水から作り出す、唯一の工業的に確立された方法として注目されている。さらに、二酸化炭素ガスによる地球レベルでの環境汚染の進行を考えると、原子力発電のロードレベリング用としての夜間の水電解、太陽エネルギーを電気に代えたのち、電解して水素として貯えるエネルギーシステムは今後重要性を増して売ると思われる。
現在工業プロセスとして採用されている水電解はアルカリ水溶液中で行われており、反応は次の通りである。
アノード反応 2OH⁻→1/2O₂+H₂O+2e (5.11)
カソード反応 2H₂O+2e→H₂+2OH⁻ (5.12)
全反応 H₂O→H₂+1/2O₂ (5.13)
理論分解電圧
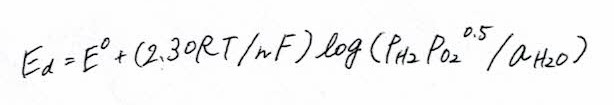
(5.14)
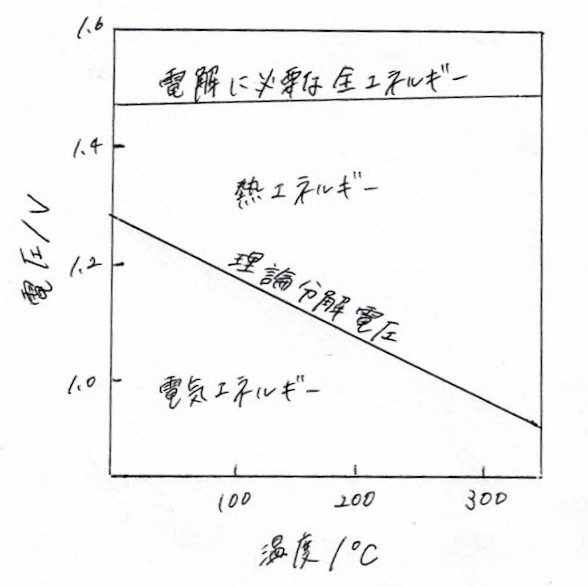
ここで、PH₂、PO₂は水素、酸素の分圧、aH₂Oは水の活量である。E⁰は標準理論分解電圧であり25℃、1atmで1.23Vとなる。しかしながら水の分解反応は吸熱反応であるため、実際に水電解を続行させるためには熱の補給も必要である。ふつうこの熱エネルギーは電気分はい時の反応の非可逆的な部分、つまり電極の抵抗や液抵抗によるオーム損失、電極過電圧によって生ずる発熱で補われる。図5.3には水の理論分解電圧(自由エネルギーにより計算される)と熱を含む電気分解に必要な全理論電圧(エンタルピー変化により計算され、理論稼働電圧(enthalpic voltage)とも呼ばれる)の温度依存を示す。電解に必要な全エネルギーは温度が変化しても大きなちがいがないか、高温になると熱の形で使用できる部分が大きくなり、理論分解電圧が低下することがわかる。
電解質としては、酸での電解も可能であるが、鉄あるいはニッケルと共存性ノ良いアルカリ水溶液が用いられる。20~30%KOH水溶液が用いられるが、ここでは電解質の抵抗を下げる点に留意されている。
電極材料はアノードにはニッケルメッキを施したスチール、カソードには軟鋼をそのままかニッケルメッキで安定したり、硫化処理して活性を上げて使用されている。いずれの電極も表面を粗面化して活性化し、ガス抜けを良くするため基本的にはエクスパンドメタルなどを使用している。
生成する水素、酸素の分離のための隔膜には石綿膜が用いられており、ニッケル線などで補強されることもある。電解槽温度は電極過電圧、液抵抗の面から考えて高温が望ましく、80℃位で運転される。この温度は材料として鉄が使用できる上限で、これを超えると個々の装置材料の劣化が著しく、新たな材料の開発が必要となる。
表5.5には現在運転されている水電解槽も諸元を示す。圧力は中将は常圧であるが、Lurgiの方式だけが実用化している中では唯一加圧式である。
| Electrolyzer Corp | BBC | Norsk Hydro | De Nora | Lyrgi | |
|---|---|---|---|---|---|
| セル方式 | 単極 | 複極 | 複極 | 複極 | 複極 |
| 圧力(kg/cm³) | 常圧 | 常圧 | 常圧 | 常圧 | 30 |
| 温度(℃) | 70 | 80 | 80 | 80 | 90 |
| 電解液(KOH,%) | 28 | 25 | 25 | 29 | 25 |
| 電流密度(A/m²) | 1,340 | 2,000 | 1,750 | 1,500 | 2,000 |
| 槽電圧(V) | 1.90 | 2.04 | 1.75 | 1.90 | 1.86 |
| 電球効率(%) | >99.9 | >99.9 | >98 | 98.5 | 98.75 |
| 電解電力(kW・h/Nm3・H₂) | 4.9 | 4.9 | 4.9 | 4.6 | 4.5 |
得られた水素を燃焼させる時に発生する熱量(ΔH)を投入した電気エネルギーで除した値は水電解のエネルギー変換効率(enegry conversion effciency)という。この値は現状では70~80%である。より効率の高い電解槽も開発中である。高温・高圧水電解法は高圧にして水の沸騰を防いで高温での水電解を行う。理論分解電圧、電極反応抵抗、液抵抗が下がり、さらに生成ガスの圧縮の仕事が軽減されるという利点を有するが、構造材料に問題が多い。アルカリ水溶液の代わりに電解質として水素イオン導電体である高分子固体電解質(solid polymer electrolyte,SPE)を用いた新電解法はSPE電解と呼ばれる。これはSPE膜上に直接電極をつけるので、コンパクトな槽構造となり、高電流密度での操作が可能となる。小型のものは商品化されている。
食塩水を電解すると、塩素ガス、水酸化ナトリウム(苛性ソーダ)、それに水素が得られる。 これは工業的に食塩電解、ソーダ電解、あるいは塩素・アルカリ電解と呼ばれる。 食塩電解は基幹産業として重要な位置にあり、電気エネルギーの消費量も 我国では電解工業の中で最も大きい。 食塩電解プロセスとしては、後述する通り、隔膜法、水銀法、およびイオン交換膜法の3つがあるが、 このうち水銀法は我国においては水銀の環境の影響を考慮して1986年をもって廃止された。※
(1)電解採取
原鉱石を必要に応じて予備処理を行ってから、硫酸などの適当な溶媒を用いて目的金属を抽出し、不純物を分離、精製したものを電解浴に入れ、電気分解を行い、カソード上に目的金属を析出させ、採取することを電解採取という。技術的には目的金属よりイオン化傾向が小さいか、ほとんど同程度の金属イオンはなるべく浄化して分離する必要がある。これらが溶液中に残存しているとカソードに析出し、製品の純度を低くする。 工業的なプロセスでは電解質は水溶液か溶融塩に限られる。後者については別項で取り扱うので、ここでは水溶液電解についてのみ示す。我国では亜鉛、カドミウム、ニッケル、コバルト、マンガン、クロムが主な製品であるが、なかでも亜鉛の 規模が大きい。 亜鉛は黄銅などの台金材料、ダイカスト材料、マンガン乾電池の負極材料としても使用されているが、最も大きい用途にトタン板で代表される鉄鋼の防食被覆がある。これは亜鉛の表面に耐食性のある塩基性被膜を生じ易いこと、亜鉛のイオン化傾向が鉄より大きいことを利用したものである。 この亜鉛をつくる方法には乾式法と湿式法がある。原料の亜鉛鉱石は硫化亜鉛 (ZnS)が主成分である。いずれの場合も鉱石を焙焼により空気酸化して酸化亜鉛とする。
乾式法とは酸化亜鉛を炭素から生成する一酸化炭素の還元力を利用して還元するものである。外部加熱方法と内部加熱方法とがあるが、高温で生成した金属亜鉛蒸気の再酸化を防ぐため、溶融鉛蒸気のシャワーで急冷する方法がISP法と呼ばれ、主流である。 湿式法とは電気分解により還元する方法で、焙焼で生成した酸化亜鉛を次のような行程で処理する。
電解の際、カソード反応が亜鉛金属の析出反応、アノード反応が酸素発生反応となる。カソードのスターティングシートとしては析出亜鉛剥離性のよいアルミ板が、アノードとしては鉛あるいは鉛合金が使用されている。電解法の特徴として大量生産には不向きであるが、小規模生産には、取り扱いが容易である点を含めて適した製法となる。 表5.7には主要な電解採取プロセスの例を示す。電解液としては硫酸塩が主体であるが、カソード生成物の表面性状が良好であることが必要で、にわかなどの添加剤が入れられる。
| 金属 | Zn | Co | Mn | Cr |
|---|---|---|---|---|
| 電解液 | ZnSO₄+H₂SO₄ | CoSO₄+H₂SO₄ | MnSO₄+(NH₄)₂SO₄+H₂O | NH₄Cr(SO₄)₂+H₂SO₄ |
| 温度 | 40 | 55 | 35~40 | 53 |
| 槽電流 | 42 | 10~16 | 7.5 | 16 |
| 電流密度 | 80~480 | 125~200 | 270 | 750 |
| 単槽電圧 | 3.3 | 3.5 | 4.7 | 4.2 |
| 電流効率 | 90 | 83 | 60~63 | 45 |
| 電解電力 | 3000 | 4200 | 8000 | 16500 |
| その他 | - | - | 隔膜あり | 隔膜あり |
(2)電解精製
乾式製錬で得られた粗金属を鋳込んだアノードを用い、目的金属と同一の金属塩を含む浴を電解液として電解し、カソード上に純度の高い金属を析出させる方法を電解精製という。 図5.6には銅を例とした電解精製の原理図を示す。電極の間に大きな電位差を与えなければ、原料のアノードに含まれる不純物のうち、その溶解電位が目的金属より貴(イオン化傾向が小)なものは溶解せずに アノード上に残るか沈殿物(アノードスライム)となる。このスライムの中には金、銀、白金といった効果な貴金属が含まれており、別行程で回収される。目的金属より卑な電位(イオン化傾向が大)の不純物は溶出するが、 カソードには析出せず、溶液中に残る。目的金属と溶解電位が近いものは分離が困難となるので、予め分離しておく。工業的には銅が代表的で生産量も多い。その他、水溶液電解でスズ、銀、白金などの貴金属、ニッケル、鉄、鉛、アンチモン、スズ、ビスマスなどで行われている。 溶融塩を電解質として用いたものでは三層電解法によるアルミニウムが実用化しているほか、プルトニウム、鉛、ウランなどについても考えられている。
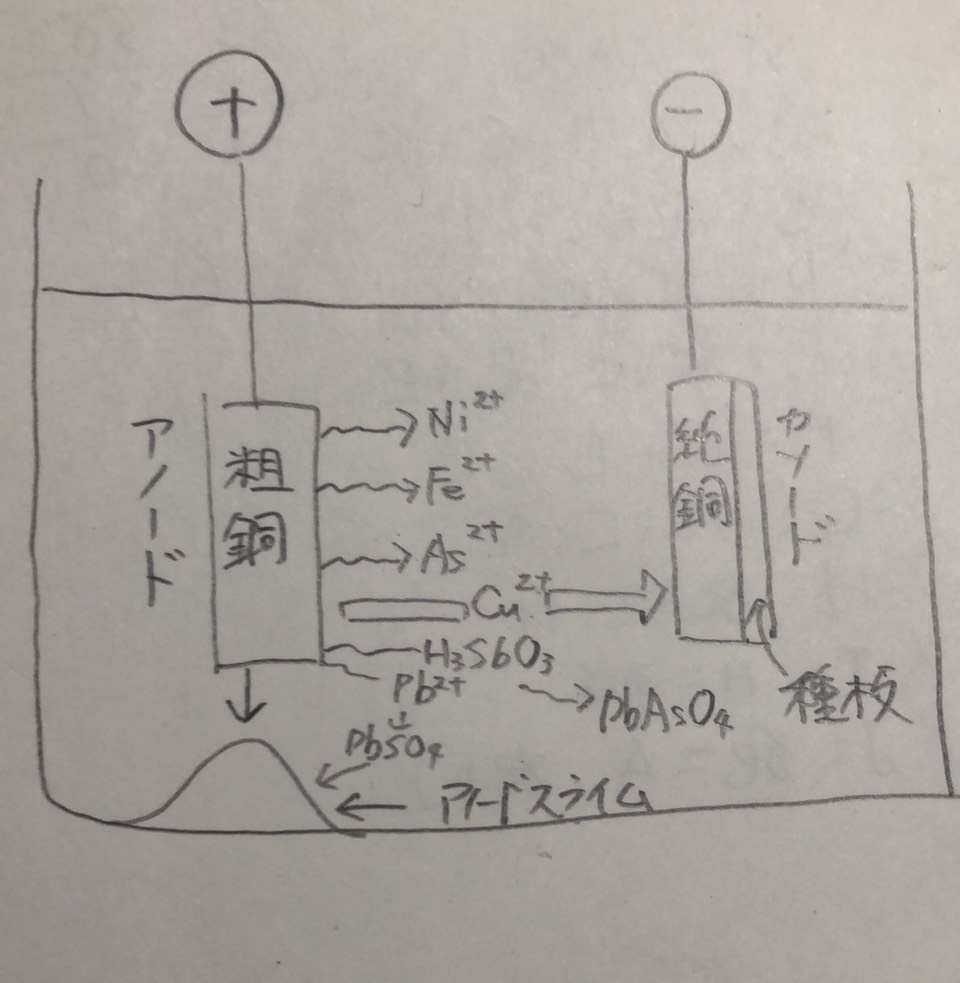
表5.8には電解精製プロセスの例を示す。目的金属がカソード上に平滑、緻密に析出することが望ましく、それに適した電解条件、電解液が選ばれる。そのために、電解液に添加剤が入れられることが多く、にかわはその代表である。
| 金属 | Cu | Pb | Ag | Au | Ni |
|---|---|---|---|---|---|
| 電解液 | CuSO₄+H₂SO₄ | PbSiF₆+H₂SiF₄ | AgNO₃+HNO₃ | HAuCl₄+HCl | NiSO₄+H₃BO₃+NaCl |
| 温度 | 60 | 常温 | 常温 | 50~90 | 60 |
| 電流密度 | 220 | 147 | 314 | 200 | |
| 単槽電圧 | 0.31 | 0.46 | 1.7 | 1.9 | |
| 電波効率 | 97 | 93 | 93 | 95~99 | 93 |
| 電解電力 | 284 | 180 | 500 | 300~400 | 1900 |
溶融塩とは常温で固体の塩を高温にして、融解させたものをいうが、イオン結晶をなす塩は融解してイオン伝導性を示し、電気化学システムの電解質となる。塩の種類により室温より1200℃の範囲で用いられるが、塩の分解電圧、蒸気圧が安定性 を評価する上で重要となる。ここでは水を使用しないのでフッ素発生のような酸素発生より貴な反応、アルミニウム還元の様な水素発生より卑な反応を起こすことができる。溶融塩電解は高温のシステムが可能で電解質の電導度が上昇するので、反 応が容易に進み、電極触媒に対する負担が小さいという利点がある。反面、高温では装置材料にかなりの制約が課されるといった欠点もある。工業電解とし は水溶液電解では製造不可能な金属の製造を中心に利用されている。リチウム、アルカリ金属、マグネシウム、カルシウムのアルカリ土類金属、アルミニウム、希土類金属、フッ素などが溶融塩電解で得られる代表的なものである。表5.9にはこれら溶融塩電解の主要なプロセスの諸元の例を示す。金属は液体の状態で生成させ、カソードとして利用したり、取り出しを容易にすることが多い。したがって浴の温度は金属の融点以上となり、それに適した塩が選ばれる。具体的にはハロゲン化物が多く用いられる。
| 製品 | Li | F | Na | Mg | Al | Ca | Mm |
|---|---|---|---|---|---|---|---|
| 原料 | LiCl | HF | NaCl | MgCl₂ | Al₂O₃ | CaCl₂ | MmO |
| 電解浴 | LiCl-KCl | KF-HF | NaCl-CaCl₂ | MgCl₂-NaCl-CaCl₂ | Na₃AlF₆-AlF₃-CaF₂ | -CaCl2 | MmF₃=LiF-BaF₂ |
| アノード材料 | 炭素 | 炭素 | 黒鉛 | 黒鉛 | 炭素 | 黒鉛 | 黒鉛 |
| カソード材料 | 鋼 | 鋼 | 鋼 | Mg(鋼) | Al(炭素) | 鋼 | Mm |
| 温度 | 450 | 85 | 590 | 700 | 970 | 800 | 900 |
| 槽電流 | 1.4 | 5 | 38 | 100 | 200 | 1.4 | 10 |
| 単槽電圧 | 6.8 | 10 | 6.9 | 6 | 4.0 | 25 | 10 |
| 電流効率 | 80 | 93 | 83 | 80 | 90 | 50 | 85 |
| 電解電力 | 36 | 16 | 10.6 | 17 | 13.4 | 67 | 11 |
| その他 | 隔膜あり | 隔膜あり | 隔膜あり | - | - | - | - |
5.3.2 アルミニウム製錬
アルミニウムの原料はアルミナ(Al₂O₃)であり、ボーキサイトを水酸化ナトリウムで処理して不純物の酸化鉄、酸化ケイ素を除き、生成する水酸化アルミニウムを焼成して得られる。 これはバイヤー法と呼ばれるが図5.7にその流れを示す。
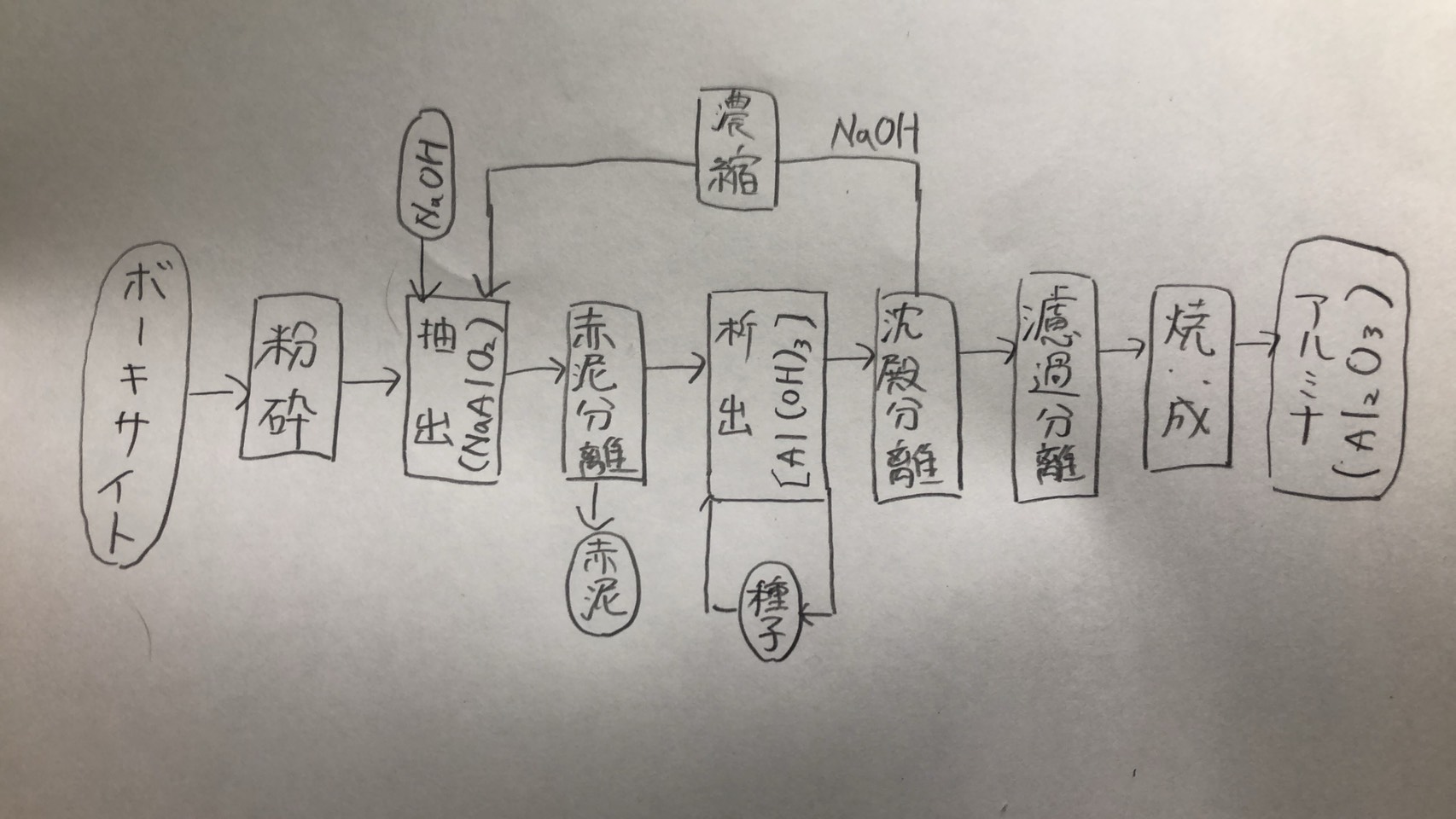
氷晶石(Na₃AlF₆)を主体とする電解浴中に、融点を下げるためAlF₃、CaF₂を添加し、970℃で、バイヤー法で得られたアルミナを5~8%溶かして電解する。これは1886年アメリカ人Hallとフランス人Heroultが別個に開発したものであるが、この開発者にちなんで ホール・エルー法と呼ばれる。反応は次の通りである。
アノードに炭素、カソードには生成したアルミニウムを利用するが、アノードの炭素が電気化学的に消費されながら反応は進む。理論的にはアルミニウム1tにつき330kgの炭素が必要だが、実際には400~450kg消費される。 この定量的に消費される炭素の補給方法に二通りある。プリベーク(既焼成)式では別な炉で炭素電極を焼成している。ゼーダベルグ(自焼成)式では電解炉に直接炭材を補給し、炉から放出される熱を利用して焼成する。 図5.8にはこれらの電解炉の構造を示す。
アルミニウムを工業的に生産する方法としては、このホール・エルー法が現状では唯一である。我が国では電力コストが高く、国内における直接生産はわずかであり、もっぱら電力の安い場所で作られる地金の輸入にたよっている。
◇イオン交換膜(ion exchange membrane)とは、イオンに解離する官能基を持っている高分子の膜のことをいい、イオンの選択透過性を有することが多い。フッ素系の膜を利用したイオン交換膜食塩電解については前に示した通りである。
通常の炭化水素系の膜の骨格構造には、スチレンー自ビニルベンゼン共重合体やスチレンーブタジエン共重合体がよく用いられる。陽イオン交換膜にはスルフォン基など、陰イオン交換膜には第4級アンモニウム基が主として導入されている。このような官能基は電解溶液中でイオン濃度やイオン種の違いにより、対イオンを解離したり、交換したりする。したがって原理的には、陽イオン交換膜では陽イオンだけが陰イオン交換膜では陰イオンだけが通過できる。イオン交換膜は、このようにイオンを通す固体電解質である。
開発初期のイオン交換膜は薬品に対する耐久性がなく、イオンの選択率も悪く、実用にはならなかった。しかし、開発の進歩により、次第に実用化されるプロセスが多くなってきた。表5.10にはその応用例をいくつか示す。
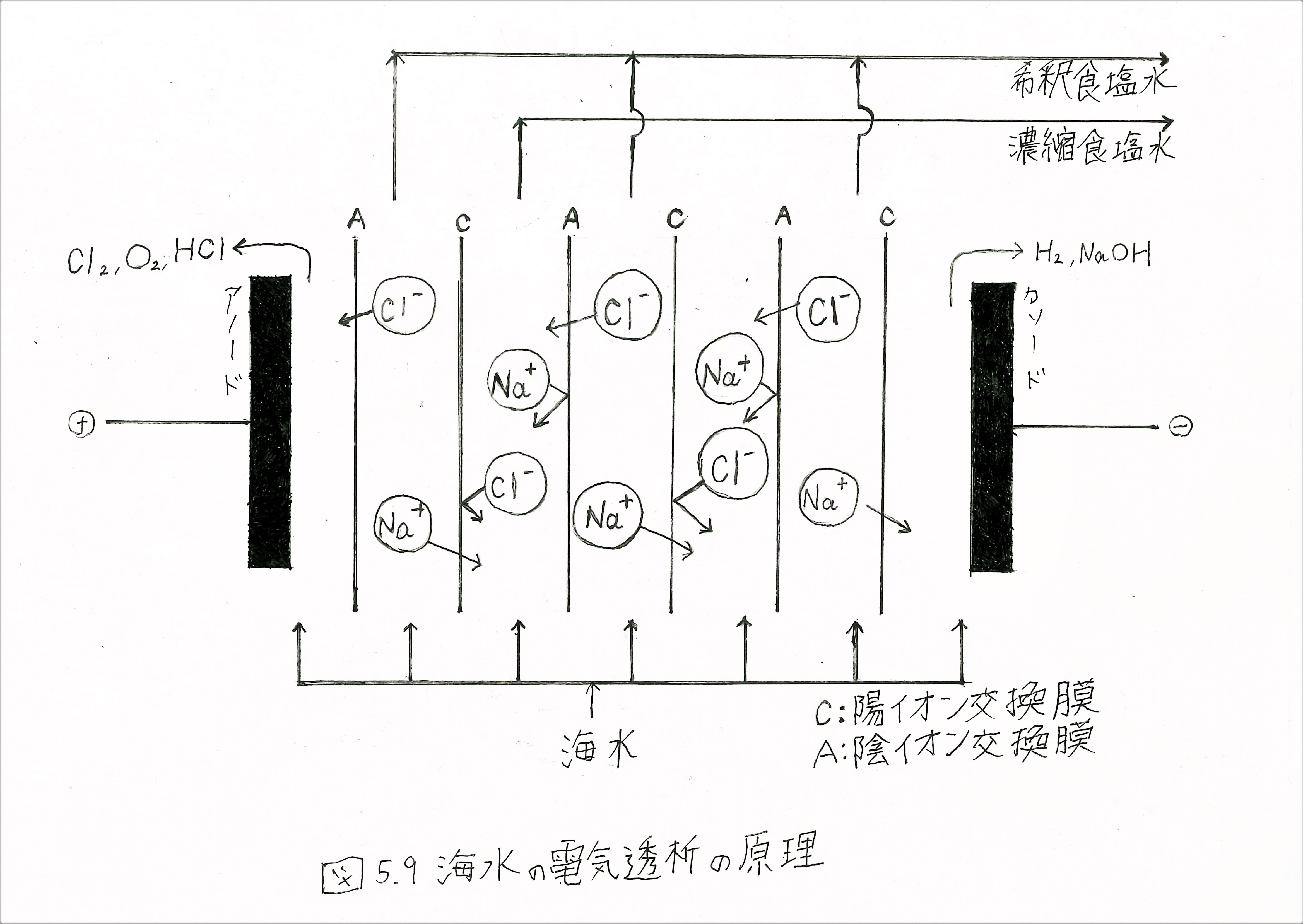
水溶液中の円の濃縮や脱塩は陽イオン交換膜と陰イオン交換膜を組み合わせて行うことができる。海水中の食塩濃度の原理を図5.9に示す。陽イオン交換膜と陰イオン交換膜が交互に並べられ、その両端に直流電圧が加えられる。電位勾配に従って陽イオンNa+はカソードへ、陰イオンCl-はアノードへ移動しようとする。ところがNa+イオンは陰イオン交換膜を、Cl-は陽イオン交換膜を通過できない。膜をはさんで塩化ナトリウムの濃い液と薄い液が交互にできる。したがって塩化ナトリウムの濃度の濃縮と脱塩が同時に行われることになる。この操作をくりかえすと濃縮度、希釈度を上げることが可能になる。
この方法は電気透析(electrodialysis)と呼ばれており、我国で行われている海水の濃縮は、すべてこの方法を経由している。食塩は濃縮液を蒸発させて作られる。かいすいを100~200ppm程度の塩濃度に希釈して工業用水とする際も、この電気透析法が用いられる。
乳児用の粉ミルクの脱塩も電気透析法で行われている。粉ミルクは、チーズ製造の副産物であるチーズホエーから作られている。チーズホエーは母乳に近い組成であるが、塩分が多く、そのままでは実用に適さない。チーズホエーに電気透析を適用すると、脱塩だけでなく、乳児に最も有害な法シュア性同位体元素のストロンチウム90、セシウム137をほかの元素より効率よくのぞけるという利点もある。
イオン交換膜は電解の際のアノード液とカソード液の混合を防ぐ隔膜としても有効である。また、食塩電解や事項で示す勇気電解合成ではセルの重要な構成要素となる。ウラン精製の際のウラニル塩(UO22+)のウラナス塩(U4+)への還元も、従来の水素還元法に代わり、イオン交換膜の新pにより、電解法の適用が可能になった。
医薬品、香料、農薬などはファインケミカルと呼ばれ、付加価値の高い製品である。これらは有機合成法や発酵法を駆使して合成されてきた。最近、これらのファインケミカルの合成に電解プロセスがきわめて有効であることが認められてきた。電気化学プロセスはもともとスケールメリットが少なく、小規模な生産でも比較的有利である。しかも、電圧や電流により反応が制御しやすい。高価な電気エネルギーを考えても、少量、多品種生産のファインケミカルには電気化学プロセスは本質的に適合しているといえる。
古くから知られているコルベ電解反応は有機電極反応の特徴をよく示している。
2CH3COO- → C2H6+2CO2+2e (5.29)
この反応はカルボン酸塩の溶液を電解すると、アノードでアセタートイオンが酸化され、炭化水素と二酸化炭素が生成するものである。このような反応は電気化学的酸化によって引き起こされる独特なものである。コルベ反応は電解条件により生成物が著しく異なる。酢酸水溶液中で白金電極を用いると、コルベ反応の電流効率は100%である。しかし、パラジウムアノード上では電位が低い(1.2 ∼ 1.6V vs.SCE)とホルムアルデヒドが生成する。氷酢酸中で白金、パラジウム、二酸化鉛電極を用いればほぼコルベ反応だけが進行するが、黒鉛電極上では酢酸メチルが生成する。
このように有機電解合成では、電極の種類、電解質、電解条件によって有機電解反応を制御できることを示している。さらに化学修飾電極を用いるなどして、立体選択性を発現させる試みも次々と報告されている。
大規模な工業的実施例としてはアクリロニトリルの電解二量化によるアジポニトルの製造がある。
2CH2=CHCN+2H++2e → NC(CH2)4CN (5.30)
これはアクリロニトリルに対する溶解度の大きい硫酸テトラエチルアンモニウム[(C2H5)4N]2SO4 などを支持電解質として使用し、pHを7.5 ∼ 9.5に調整して電解を行う。カソードで二量化が起こり、アノードでは酸素発生反応が起こる。従来は陽イオン交換膜を用いて、カソード生成物がアノード室へ混入するのを防いでいたが、近年には無隔膜の方式も開発された。
この他に、合成潤滑油や高級可塑剤として使用されるセバシン酸、除草剤のビピリジニウム塩は電解合成によって作られている。今後は付加価値の高い医薬品、香料へも電解合成法の適用が進められていくものと思われる。有機電解合成の特徴としては次の点が挙げられる。
電解プロセスでは熱のみの化学反応では容易に起こらない反応を起こすことができる。そのため、いくつかの無機化合物の製造に適用されており、狭い意味で電解酸化(electrolytic oxidation)、電解還元(electrolytic reduction)と呼ばれている。表5.11には電解により作られる無機化合物のいくつかを示す。すなわち、電解法の特徴を生かした過酸化物の製造にその例が多い。
| 製 品 | NaClO3 | (NH4)2S2O8 | K2MnO4 | MnO2 | UCl4 |
|---|---|---|---|---|---|
| 電解液 | NaCl | (NH4)2SO4+H2SO4 | K2MnO4+KOH | MnSO4+H2SO4 | UO2Cl2+HCl |
| アノード材料 | 金属電極 | 白金 | ステンレス鋼 | 鉛 | 白金 |
| カソード材料 | 軟鋼 | 黒鉛 | 軟鋼 | 鉛 | チタン |
| 温度( ℃ ) | 70 | 30 | 70 | 90 | 30 |
| 電流密度(A/m3) | 2000 | 8000 | Ja=70 Jc=700 | 100 | 1200 |
| 単位電圧(V) | 3.3 | 3.8 | 3.0 | 2.2 | 5 |
| 電流効率(%) | 93 | 78 | 60 | 93 | 83 |
| 電解電流(kW・h/t) | 5400 | 1100 | 850 | 1700 | 1850(AC) |
| その他 | - | 隔膜 | - | - | 陽イオン交換膜 |
塩素酸ナトリウム NaClO3 は食塩の無隔膜電解により作られる。食塩電解により生成した塩素と水酸化ナトリウムが溶液中で反応して、塩素酸ナトリウムが生成する。次亜塩素酸の解離定数の値を考慮し、pH=7 の中性付近で行われる。
電解反応 6NaCl+6H2 ⇄ 3Cl2+6NaOH+3H2 (5.31)
溶液内反応 3Cl2+6NaOH ⇄ NaClO3+5NaCl+3H2O (5.32)
全反応 NaCl+3H2O ⇄ NaClO3+3H2 (5.33)
塩素酸カリウム、臭素酸塩、ヨウ素酸塩も同じような反応で作られる。
過硫酸アンモニウム (NH4)2S2O8は濃厚酸性硫酸塩のアノード酸化により作られる。
2(NH4)HSO4 ⇄ (NH4)2S2O8+H2 (5.34)
電解質としては、硫酸-硫酸アンモニウムの混合液を用い、電流効率を上げるには低温にすることが望ましい。電流密度が高い方が効率が良く、アノードとしては平滑の白金が用いられる。
乾電池の正極活物質として欠かせない電解二酸化マンガンは硫酸マンガンの電解酸化により作られる。
MnSO4+2H2O ⇄ MnO2+H2SO4+H2 (5.35)
マンガン鉱を硫酸により浸出し、石灰石、硫化水素で中和、浄液を行い電解槽供給液とする。電解は低電流密度で行い、アノード上に二酸化マンガンを析出させる。乾電池材料としての特性は析出条件や後処理条件で微妙に変化する。
代表的な表面処理法と、その目的ならびに用途を示すと表6.1のようになる。いずれの処理法も表面を美しくするため(耐食・耐摩擦性化)、 あるいは特殊な機能を持たせるため(機能化)に応用されていることがわかる。したがって本章では、これらの項目に分けて述べていくことにする。
| 表面処理法 | 目的 | 用途(具体例) |
|---|---|---|
| 電気めっき | (イ)装飾 (ロ)耐食 (ハ)耐摩耗 (二)機能 | |
| 無電解めっき | (イ)装飾 (二)機能 | |
| 気相めっき | (ロ)耐食 (ハ)耐摩耗 (ハ)機能 | |
| エッチング | (二)機能 | |
| アノード酸化 化成 | (イ)装飾 (ロ)耐食 (ハ)耐摩耗 (二)機能 | |
| 電解研磨 | (イ)装飾 (ロ) 耐食 (二)機能 | |
| 化学処理 | (イ)装飾 | (イ)装飾品(カラーステンレス) |
| 泳動電着 | (イ)装飾 (ロ) 耐食 (二)機能 | |
| 表面硬化
|
(ロ)耐食 (ハ)耐摩耗 | (ロ)+(ハ)耐食・耐摩耗品(工具) |
| 化学修飾 | (二)電極の機能化(電池用電極、センサー) | |
| 電鋳 | (イ)装飾 (二)機能 |
| 表面処理法 | 目的 | 用途(具体例) |
|---|---|---|
| 電気めっき | (イ)装飾 (ロ)耐食 (ハ)耐摩耗 (二)機能 | |
| 無電解めっき | (イ)装飾 (二)機能 | |
| 気相めっき | (ロ)耐食 (ハ)耐摩耗 (ハ)機能 | |
| エッチング | (二)機能 | |
| アノード酸化 化成 | (イ)装飾 (ロ)耐食 (ハ)耐摩耗 (二)機能 | |
| 電解研磨 | (イ)装飾 (ロ)耐食 (二)機能 | |
| 化学処理 | (イ)装飾 | (イ)装飾品(カラーステンレス) |
| 泳動電着 | (イ)装飾 (ロ)耐食 (二)機能 | |
| 表面硬化
|
(ロ)耐食 (ハ)耐摩耗 | (ロ)+(ハ)耐食・耐摩耗品(工具) |
| 化学修飾 | (二)電極の機能化(電池用電極、センサー) | |
| 電鋳 | (イ)装飾 (二)機能 |
めっきといえば、あたかも装飾のみ用いられているような印象を与えるが、耐食・耐摩耗性強化のために、数多く用いられている。一方、基板を反応させて上に耐摩耗性 が高く、しかも耐食性も高い金属の窒化物・炭化物等をコー ティングする方法は気相めっきと呼ばれているが、電気めっき、無電解めっきと合わせ、この部類に入れることにする。
気相めっき 気相めっきは、一般にはCVD*(Chemical Vapor Deposition:化学蒸着法)を意味し、TiC、TiN膜形成など、特に耐摩耗性、耐食性を目的としたコーティング技術として発展してきた。しかし、近年半導体用技術として飛躍的発展を遂げ、さらに、減圧下でグロー放電させてプラズマを発生させ、より低温で基板上に膜を形成させるプラズマCVD法、エキシマレーザー等による光照射下で原料ガス分室を励起・分解させ、より低温でより精密な膜を形成させる光CVD法など、種々な新膜形成法が発展してきているが、ここでは、耐摩耗性・耐食性という面から、TiCおよびTiNのCVDをとり上げるにとどめる。
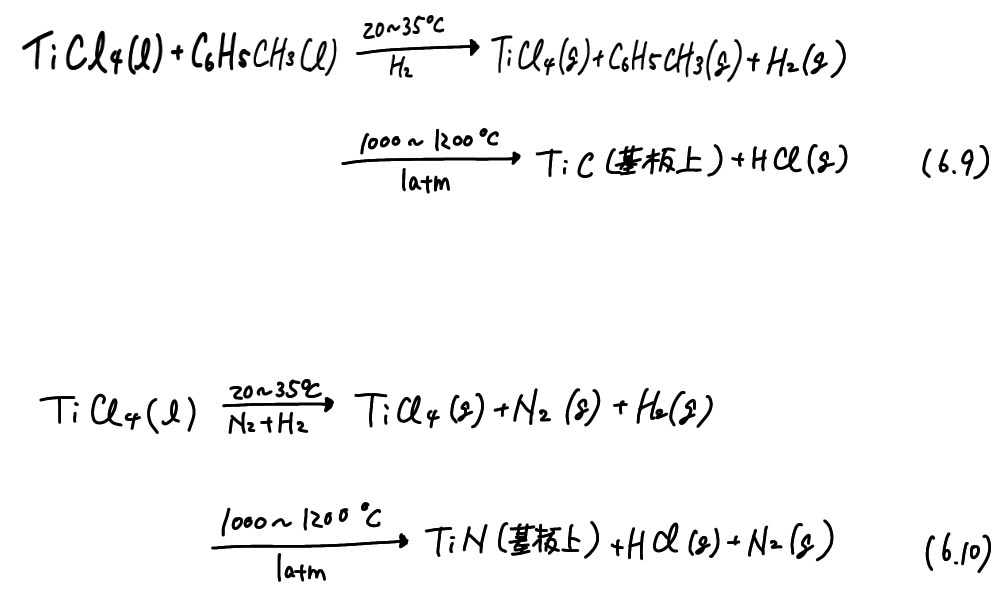
金属の炭化物、窒化物、ホウ化物、ケイ化物、酸化物などの皮膜は非常に硬度が高く、耐摩耗性に富んでいる上、TiC、TiNなどは耐食性にも優れているため、切削用工具等に広く応用されてきた。下に示すように、反応ガスの熱分解により基板上にTiCコーティングやTiNコーティングが得られる。熱CVDと呼ばれるゆえんである。
*プラズマCVD、光CVD等と区別するため、熱CVDとも呼ばれる。
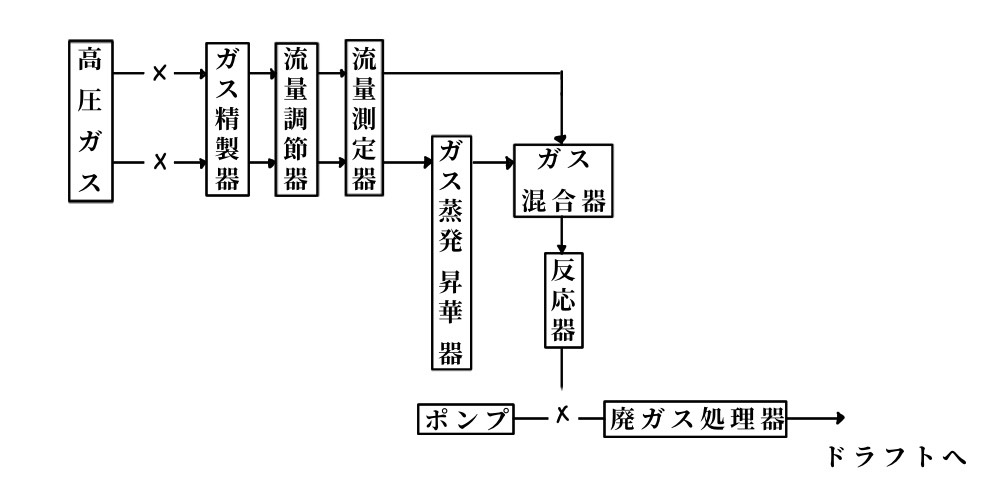
熱分解CVD装置の基本フローとシートは図6.6のようであり、加熱方法は表6.10のようで、用途に応じて使い分けられている。
電気めっき
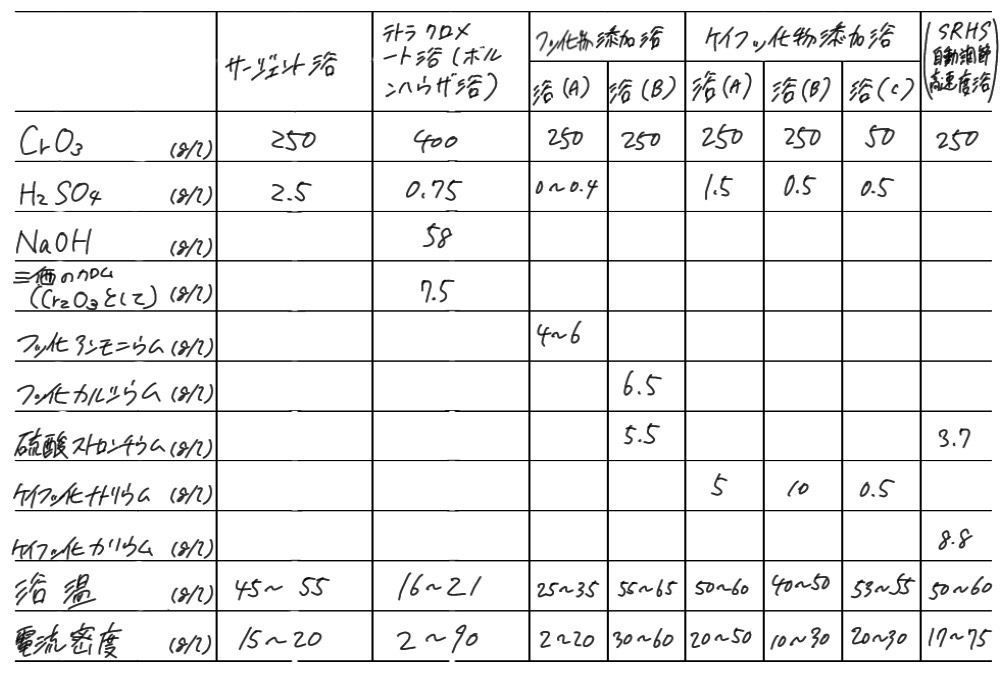
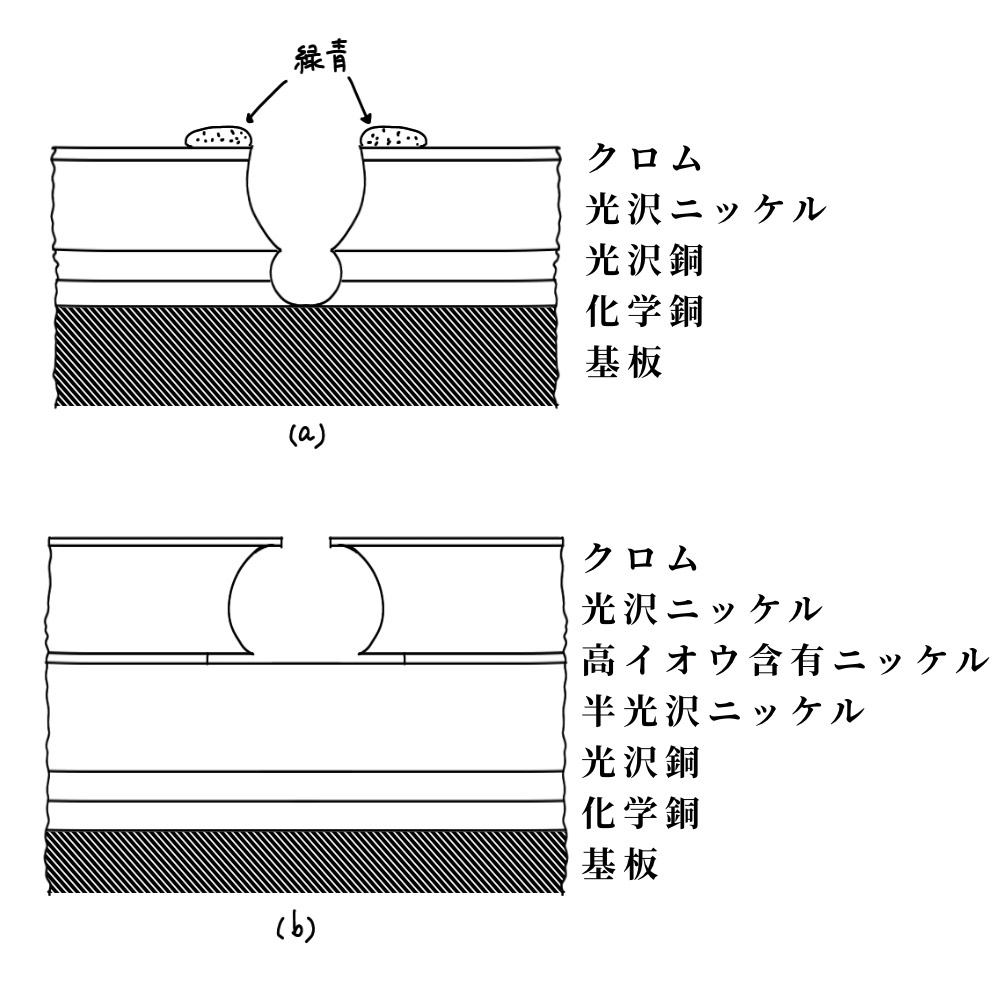
自動車のマーク等にCrめっきが施されているが、これは装飾性よりも耐候性、耐食性などに重みがおかれている。この淡青白いCrめっきには、変色せず、光沢をいつまでも失わないCrの特質が活されている。クロムめっき浴は表6.11のようで、アノードには不溶性電極として鉛および鉛合金が用いられる。光沢クロムめっき(約1μm厚)にはピンホールやごく小さな割れ目が生じやすいので、その上に約0.25μmのクロムめっきを施す二重クロムめっき法も用いられる。しかし、少しでも腐食されると下地のCuがでてきて緑青(Cuの緑色のさび)となり、ひどく外観を損ねるので、強力な耐食性をもたせるため、図6.7に示すような三重ニッケルめっきが施され、何年間も美しく光輝く自動車マークが作られる。
図6.7 めっき皮膜における腐食の模式図 (a)単層ニッケルめっき (b)三重ニッケルめっき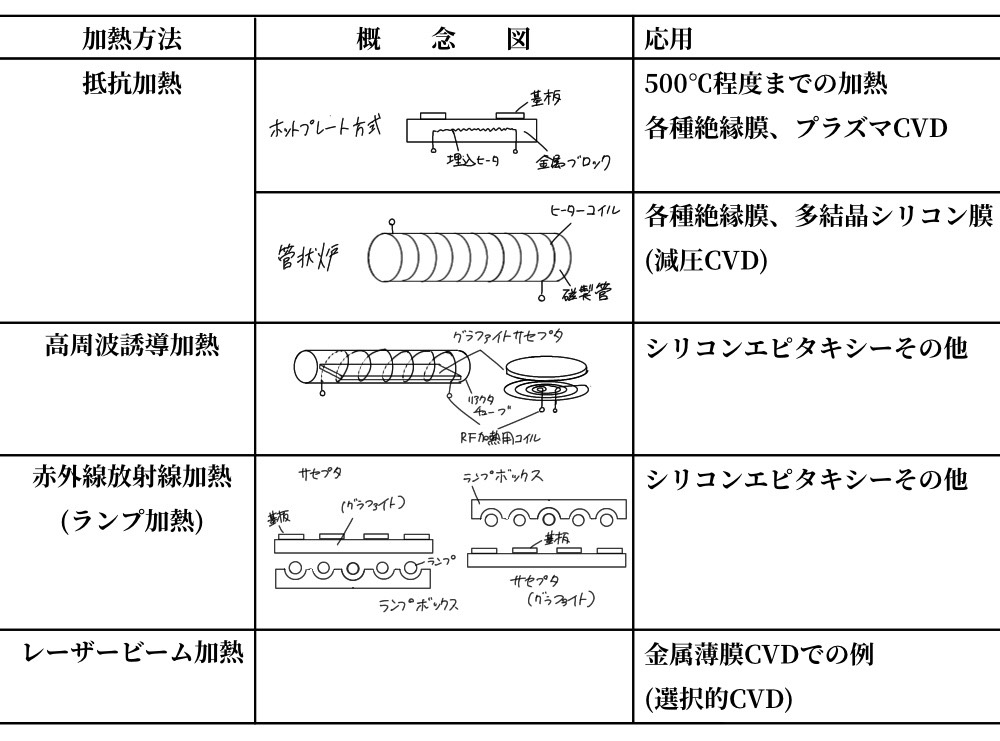
無電解めっき NiP-BN複合皮膜を無電解法により作製して、低摩擦係数の部品を得たり、無電解法によるNi-P-Cr3C2複合めっきを応用した金型を作製して、Co-Mo合金に匹敵する程の耐摩耗性を示すものを得ている。さらには、無電解法によるNi-Bをマトリックスとするダイヤモンド複合めっきなども検討されている。
6.2.2でも述べたが、Alのポーラス被膜では約200μmの膜厚のものも得られ、硬くて耐摩耗性が大きく、また緻密で密着性、絶縁性に優れている。さら着色も容易であり、熱水と反応させる封孔処理(sealing treatment)(図6.8)によって孔をふさぐことにより、皮膜の退色を抑え、耐食性・耐候性を大幅に改善することができる。 Alアノード酸化皮膜の機能および応用は表6.12のようで、耐食性・耐候性のみにとどまらず、幅広い応用が可能である。
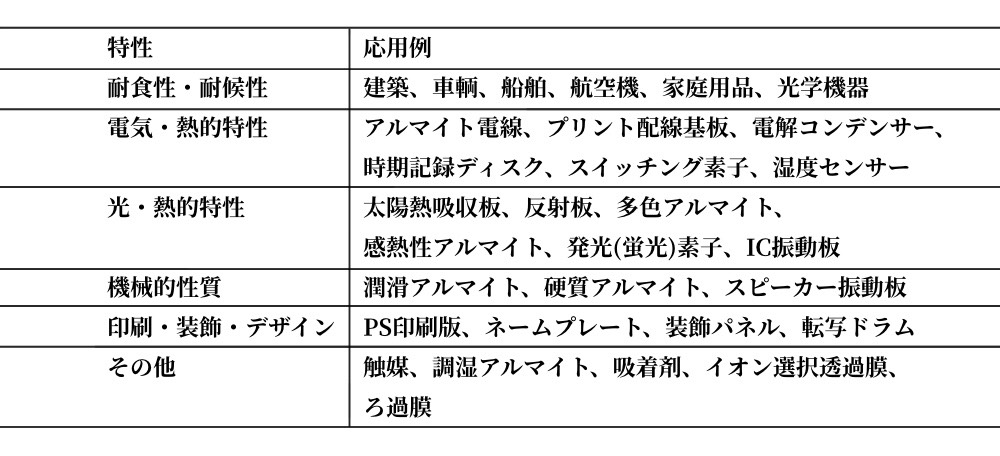
注)現在研究・開発中のものも含む。
また、純度の高いジルコニウムは、原子炉において、ウラン燃料を包む金属材料として重要である。原子炉内には冷却水が循環しており、この水による腐食を防ぐため、ジルコニウムをアノード酸化して、非常に耐食性の強い防食皮膜を形成させ、使用している。
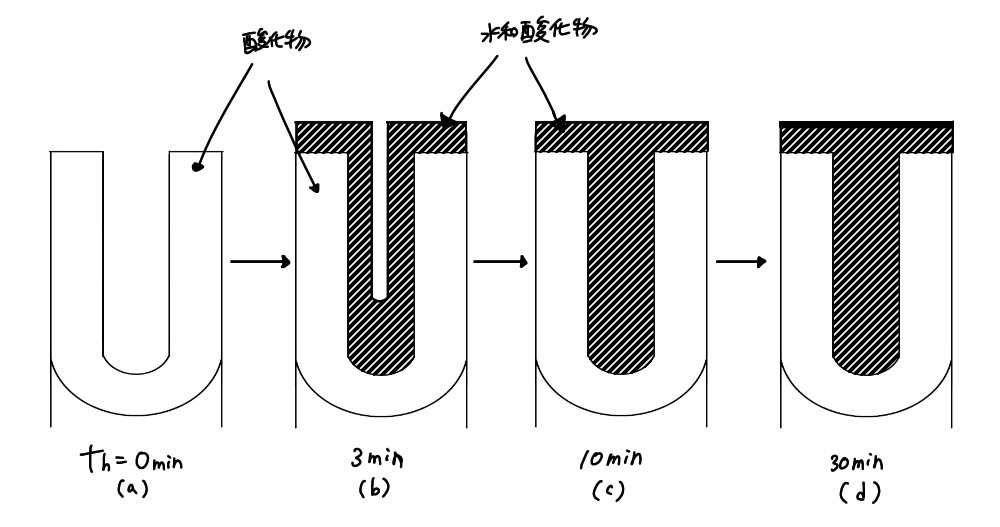
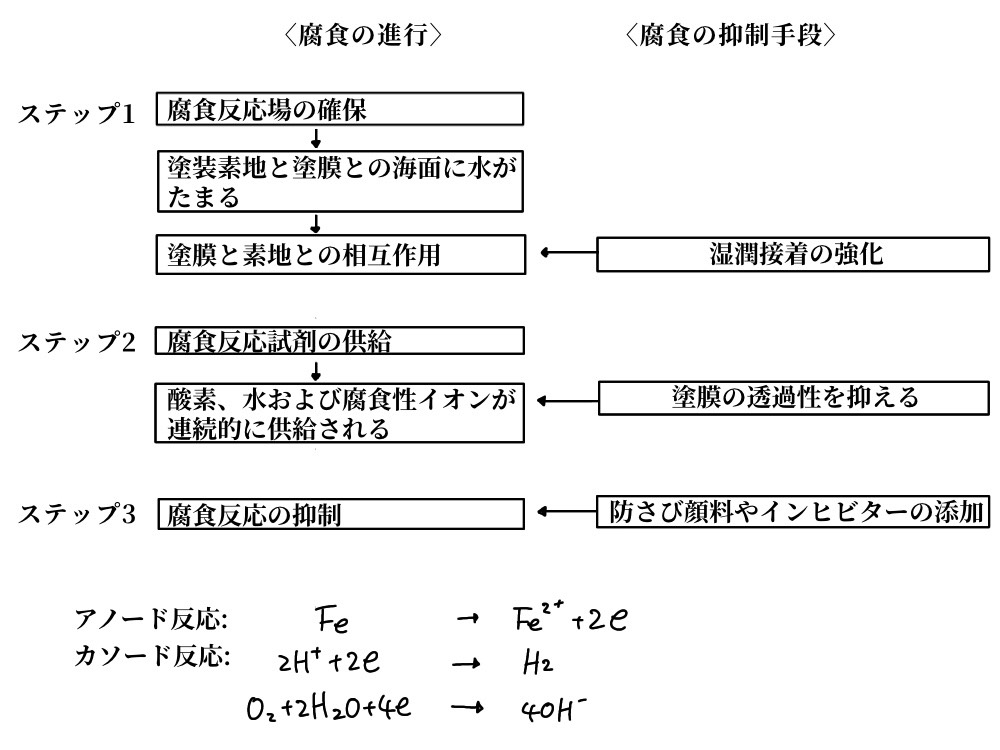
塗装の防食機能は、外界の腐食性物質と素材との接触を阻止する遮断機能と、防 食顔料やインヒビター(inhibitor)による腐食反応抑制機能にある。遮断機能では、全際と塗装素材との相互作用に大きく依存する湿潤接着が重要である。そこで、塗膜下の腐食の進行を図6.9に示すような三段階で考えた場合、①Step1は湿潤接着力の強化で、②Step2は塗膜の透過性を抑えることで、③Step3は防さび上顔料やインヒビター(腐食抑制剤)を添加することで、それぞれ抑制することができる。すなわち、これらの機能を塗料(塗装)に持たせれば耐食性は十分といえるわけで、①、②は下地塗装に、③はいわゆる塗装に機能させている。後者については、6.2.3で述べたので、ここでは自動車ボディーの塗装の下地処理として行われる電着塗装(electrodeposition coating)について述べる。
電着塗装 本法は、電着浴内に浸した被塗物と対極とのあいだに電圧をかけると、水の電気分解によって電極近傍の界面pHが急変し、そのため塗料粒子が凝縮・付着する特質を利用する塗装技術である。塗料は高分子電解質で、その電荷の正負に従ってカソードあるいはアノードに電着するので、それぞれカチオン電着塗装あるいはアニオン電着塗装と呼ばれる。塗料が電着した部分は皮膜抵抗となって分極増大して電着しにくくなるため、電着は未塗装部分に順次移動して、つきまして性が非常に良くなるとともに、皮膜内では電気浸透が発生して内部の水を移動して脱水を行結果、被装物全体が緻密で均一な塗膜で覆われることになる。アニオン電着では、被装物をアノード分極するため素地金属の溶出を招く。そのため、防食基準の厳しい自動車の車体の塗装では、ほとんどカチオン電着塗装が用いられている。カチオン電着塗装では、エポキシウレ タン、アクリルウレタンなどポリアミン樹脂を低級有機酸で部分的に中和・解膠した塗料を使用している。通常、印加電圧150>400V、温度25~30℃、膜厚20~25μmの場合、2~ 3分で塗装が終了する。電着塗装過程は表6.13ようにまとめられる。

ここでは、基材表面にC、Nなどの非金属、あるいはAlなどの金属を拡散させて表面に拡散皮膜(diffusion film)を形成させ、腐食、特に高温腐食を抑制するとともに、表面を硬化させて対摩耗性を向上させる技術をとり上げる。
表6.14 各種表面熱処理を施した銅(0.40%C)の 摩擦摩耗特性(GREGORY)
窒化処理 この窒化(nitriding)処理法も、西ドイツより塩浴軟窒化法(タフ トライド法)が導入されて以降、急激な発展を遂げた。NH3に少量の酸素あるいは空気を添加した雰囲気中で鋼の処理を行う酸窒化処理法や、耐摩耗性、耐焼きつき性の改善に、表面に生成された硫化物の存在が効果的であることを考慮した、窒化性塩浴にKCNSやK2S等を少量添加した浴中で、570℃前後で鋼を処理する浸炭窒化処理法などがある。これらの効果は、表6.14を見れば明らかである。
浸透処理 表面硬化を目的とした浸透処理としては、主としてAl、Cr、Ti、Wなどの金属、またはそれらの炭化物、さらにS、Bなどを金属基板表面から拡散浸透させる方法がある。これらのうち、Al、Crについては、鋼材等の耐熱・耐食性を向上させる処理法としてのアルマイジング(カロライジング)、クロマイジングが古くから知られ、工業化されてきた。さらには、溶融ホウ砂中、あるいは2KCI・BaCl2溶融塩中にTiなどの拡散元素をK2TiF6十Ti等の形で溶融させ、鋼中の炭素と反応させて、鎖表面に非常に高硬度なTiC層を形成させる方法、B4C(80wt%)十Na2CO3(20wt%)から、1000℃において鋼表面にBを補給してFe2B層を形成させる方法などが開発されている。
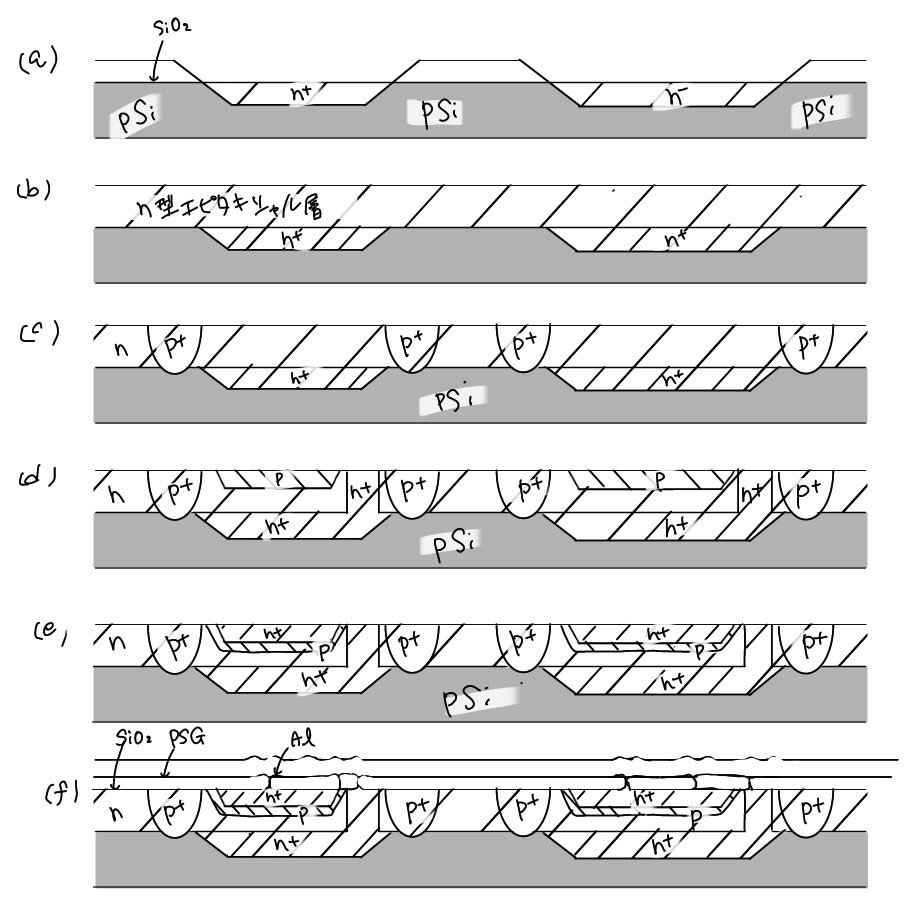
半導体上pn接合、絶緑膜金属を組み合わせて、トランジスクギダイオードなどの素子を作製し、目的にかなった回路を作っていくのがJICのプロセス技術である。この製造法を可能した基本的な技術は、Siへの不純物拡敵技術とプレーナー技術である。プレーナー技術はSi表面上にSiO2を形成しフォトリソグラフィー投術を利用して不必要な部分のSiO2を取り除いて、不純物の拡散を行い、さらにこの工程を繰り返して、デバイスを構成する方 法で ある。次に各々の素子間の分離、起線などが行われ、1つのICができ上がる。したがって、1つのデバイスを作製するに は、Ⅰ)リソグラフィー、Ⅱ)エッチング、 Ⅲ)拡散、Ⅳ)成膜の各技術が必要となり、これらの技術が総合的に利用されることにより、ICデバイスが実現するわけである。ここで、具体的にプロセスを理解するために以下にベイボーラICとnMOS-IC作製プロセスを概略する。
バイポーラICの製造プロセスを図7.8に示す。バイポーラICでは、名素子間を電気的に絶縁する分離技術が主要技術となっている。基板としてのp型ウェハにまず野酸化によりSiO2膜を作製する。フォトレジストの塗布、アライナーによるパ
MOS-IC はバイポーラに比して構造が簡単で製造工程も少ない。MOS-ICは、初期にはpチャンネル型のpMOS-ICが利用されたが、IC製造技術が進み、nチャンネルのnMOS-ICが主に利用されるようになっている。nMOS-ICは電子キャリヤーであるので、pMOS-ICより高速で作動する。最近ではnMOSとpMOSの両トランジスタを含むcMOS-ICが、低消費電力の制御、信頼性の点から重要となっている。
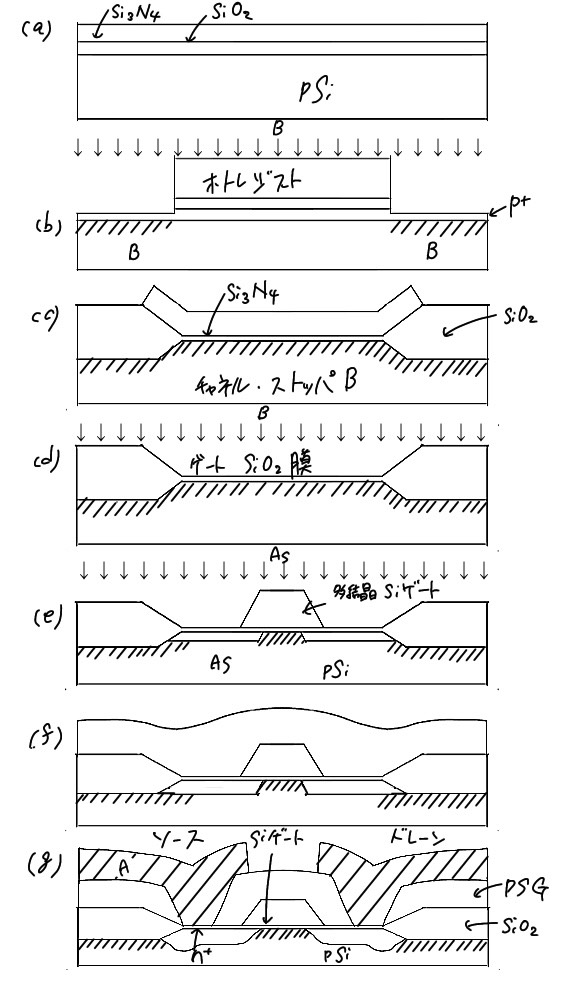
図7.9にnMOS-ICの製造プロセスを示す。不純物としてホウ素を拡散したp型Siウェハ表面をわずかに酸化し、SiO2とした上にシリコン窒化物(Si3N4)をCVDにより堆積する(a)。 次にフォトリソグラフィーによりSi3N4膜を選択エッチングし、ホウ素のイオン注入を行う(b)。 ホウ素は不活性領域にのみ打ち込まれる。これは、p領域表面がn型に反転するのを防止して電気的絶緑を保ち素子間分離を行っている。次にレジスト除去後、熱酸化によりSi3N4膜のない部分に厚いSiO2膜を形成する(c)。 この上を熱リン酸処理してSi3N4膜を除去する(d)。 このようにSi3N4膜を利用してSi表面を選択酸化する方法をアイソプレーナー法と呼んでいる。熱酸化により薄いSiO2膜を形成後、MOSトランジスタのしきい値電圧を制御するためにホウ素をイオン注入する(d)。 その上にCVD法により多結唱Si膜を堆積する(e)。 このときSi膜にリンを十分高測度に添加し電気抵抗を下げてゲート電極とする。次にエッチングで ゲート部分以外の多結晶Siを除去し、n+不純物(As)をイオンとして打ち込む(e)。 このAsが後の高温熱処理で再配列し、活性化され、ソースとレーン領域を作る。次に厚いPSG膜、またはPSGとSiO2複合膜をCVD法で堆積し(f)、電極用コンタクトホールをフォトリソグラフィーであける。Alを蒸着し、フォトリソグラフィーで不用部をエッチングする(g)。 普通はこの上保護膜としてSiO2やSi3N4膜を低温堆積させるとMOSトランジスタが完成する。


問題
7.1 FETの作動原理を説明せよ。
7.2 バイポーラICの特徴をモノポーラICと比較して説明せよ。
7.3 プリント配線板の電子部品材料における役割を説明せよ。
7.4. 垂直磁気記録方式を従来の長手記録方式と比較して説明せよ。
多くの生体反応は、本質的に電気化学反応であると言っても過言ではない。生態系が持っている種々の優れた生体反応を理解し、それを応用することができれば、これまでとは違った高度で新しい科学の領域が開かれることにもなる。 バイオエレクトロケミストリーは、生体系の理解とその制御・応用を目的としてバイオテクノロジーとエレクトロニクスと化学が融合した電気化学の新しい領域として拡がりつつある。
ここでは、バイオエレクトロケミストリー(生物電気化学,bioelectrochemistry)のいろいろな分野について述べる。
生体内で生じる反応は、基本的には酵素(enzyme)が触媒として働く反応と考えてよい。この酵素反応と電極上で生じる電気化学反応とは、多くの似たところがある。例えば、細胞膜(cell membrane)に埋め込まれた酵素上での反応は固相/液体界面のような異なった相の境界で起こる電極反応と同じく不均一界面という場での反応と考えられること、細胞膜も電極もどちらも電荷を持った表面であること、さらに生体内での反応のpH,イオン強度や温度などは電気化学反応の際に通常よく用いられる条件と類似していることなど、を挙げることができる。特に酸化還元を行う酵素の反応は、酸化還元という電子の移動を取り扱う電気化学の最も得意な領域であって真に電気化学反応と考えてよい。我々の生命活動の源である呼吸を通してのエネルギー生産過程、植物の光合成いずれも酸化還元反応を基礎とした電気化学反応である。
電気化学は、計測するという観点から生体反応の熱力学的な情報(例えば酸化還元電位(redox potential)、したがって電子の流れる方向)や電子が移動する速度およびメカニズムに関する情報を得る手段にもなる。膜の電位やイオンの移動に関する電気化学の基礎的な概念は、生体膜の機能を理解したり測定するための重要な基礎となっている。ライフサイエンス(生命科学、life science)の重要な領域としての生体情報の計測、変換およびその医療への応用に電気化学が果たす役割は大きい。電気化学的な方法を用いて生態系をまねた人工的なモデル系がつくられている。神経モデルや光合成モデルなどがそれである。電気エネルギーや有用物質の電気化学的生産のために酵素や微生物の利用も考えられている。
図8.1に、バイオエレクトロケミストリーの拡がりを概念的に示した。
生体内の反応は、数多くの酵素反応が組合さって進行する。酵素はよく知られているように、その特異な蛋白質の構造に由来して特定の物質とのみ特異的に反応するので生化学物質の選択的反応、合成さらには計測のためによく用いられる。さて、このような酵素反応を電気化学的に検出するにはどうすればよいのだろうか。もし、酵素反応で消費あるいは生ずる物質が電極反応をする(電極反応活性といい電極上で酸化または還元されることをいう)場合、それらを検出しさらにそれを制御することによって酵素反応が生じたかどうかを知ることができる。図8.2(a)の場合である。これはもっとも簡単な酵素反応の電気化学的検出方法で、酸素を消費して過酸化水素を生ずる反応が同時に進行する酸化酵素などの場合に用いることができる。今日ではこの方法を用いて酵素センサが作製、実用化されている。しかし、この方法は反応物や生成物が電極反応活性でない場合や酸素がない場合には酵素反応そのものが進行しないので利用できないし、酵素反応を能動的に制御したとは言い難い。電極を用いてもっと積極的に酵素のような蛋白質の電子移動をコントロールできないだろうか。次に述べる図8.2(b)および(c)はそれを目指したものである。
電気化学は、計測するという観点から生体反応の熱力学的な情報(例えば酸化還元電位(redox potential)、したがって電子の流れる方向)や電子が移動する速度およびメカニズムに関する情報を得る手段にもなる。膜の電位やイオンの移動に関する電気化学の基礎的な概念は、生体膜の機能を理解したり測定するための重要な基礎となっている。ライフサイエンス(生命科学、life science)の重要な領域としての生体情報の計測、変換およびその医療への応用に電気化学が果たす役割は大きい。電気化学的な方法を用いて生態系をまねた人工的なモデル系がつくられている。神経モデルや光合成モデルなどがそれである。電気エネルギーや有用物質の電気化学的生産のために酵素や微生物の利用も考えられている。
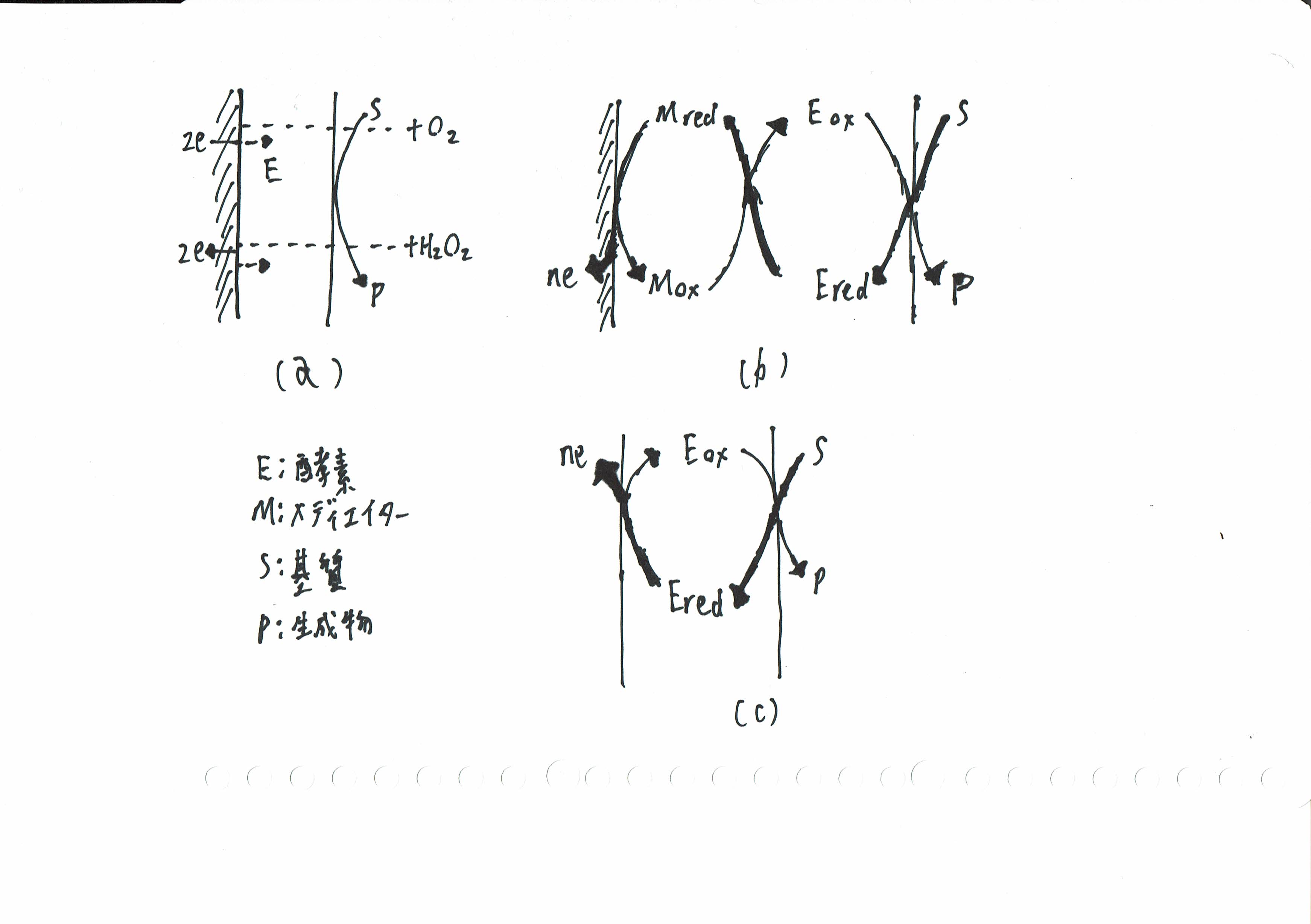
生化学物質の酸化を行う酸化酵素を考えてみよう。酵素は活性中心と呼ばれる酸化還元にかかわる部位が蛋白質に取り囲まれた構造をとっている。酵素からその活性中心を取り出すと、一般に電極上で電気化学的に酸化還元を行なうことができる。しかし、蛋白質と一体となった酵素そのものの酸化還元を直接電気化学的に行うことは容易ではない。これは、酸素や過酸化水素あるいは酵素から取り出した活性中心またはそのモデル化合物の様な小さな分子は電極と直接接触できるが、これに比べて、分子サイズが大きい蛋白質では、その分子内の奥深くに埋もれた活性部位が、電極に近接できないので電極との電子のやり取りが起こりにくいためと考えられている。このような場合、酵素の活性部位まで侵入して酵素と電子のやり取りをし、電極まで運んでくれる小さな分子を仲介役として用いると、電極を用いて酵素を酸化(あるいは還元)することができる。このような電子を運ぶ役割を担う分子をメディエイター(電子移動媒体,mediator)とか電子シャトルという(図8.2(b);図8.3)
グルコースを酸化する酵素であるグルコースオキシダーゼ(GOD)の場合、例えばフェロセンやハイドロキノンのような分子をメディエイターとして用いると電極で酸化されたメディエイターがGODの活性中心(基質によって還元された型になっているフラビン部位)から電子を受け取り、電極まで運んできたのち、電極上で再び酸化される。こうして、酵素と電極との間で電子のやり取りができることになる。
ここで、どんな分子でもメディエイター分子になれるわけではない。各々の酵素にはそれぞれに適したメディエイター分子がある。GODと同じフラビン部位を活性中心とするフラビン蛋白質でも、アミノ酸であるサルコシンの酸化酵素や乳酸の酸化酵素は、それぞれGODに有効なメディエイターよりもむしろモリブデンやタングステンのシアノ錯体が有効なメディエイターとなることが知られている(図8.4)。適切なメディエイターを捜すことは必ずしも容易ではないが、メディエイター分子の酸化還元電位、分子の形、親水性・疎水性、電荷状態などが重要で、おそらく酵素の活性中心に接近でき、電子授受を行うのに適した条件が要求されるのであろう。酵素は適切なメディエイターがあれば、電気化学的に酸化(あるいは還元)できるので(この場合酸素がなくても働く)これを利用したセンサや酵素反応を利用した合成反応用電極を作ることができる。また、電気化学の手法を利用して酵素反応の過程を解析し、酵素反応を特徴付けるミカエリス定数や反応速度を求めることもできる。すなわち、一般にメディエイターを用いた溶液内での酵素反応速度(v)は、
v=k[E]/(1+KS/[S]n+KM/[M]n)(8.1)
ここでkは速度定数(s-1)でvmax=k[E]、KSおよびKMはそれぞれ基質およびメディエイターに対する酵素反応のミカエリス定数(酵素と基質の複合体の解離定数=[E]・[S]/[ES]に相当し、酵素反応の最大速度vmaxの1/2の速度の時の基質濃度)、[E]、[S]、[M]は酵素、基質、メディエイターの濃度を表す。nは通常1(または2)である。図8.4(c)の場合の電流がどれだけ大きくなるかを種々の条件下で測定することによって上記のパラメーターが評価できる。このようにメディエイターを用いることで、酵素反応を外から電気化学的に制御することができるようになる。